Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Genetics. 2012 Oct;192(2):651-69. doi: 10.1534/genetics.112.139519. Epub 2012 Jul 30.
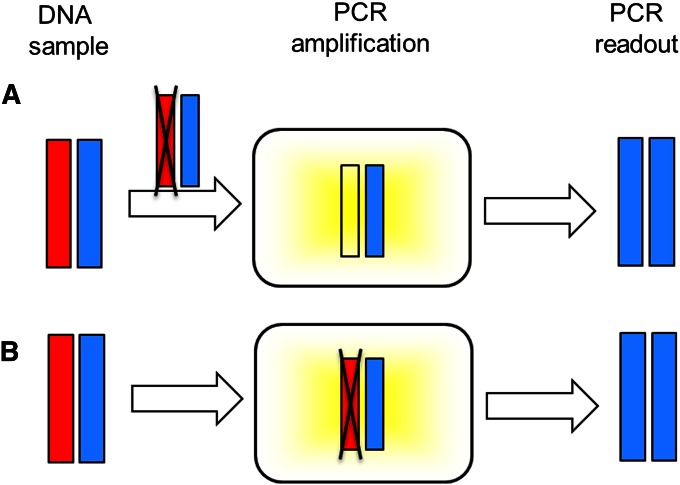
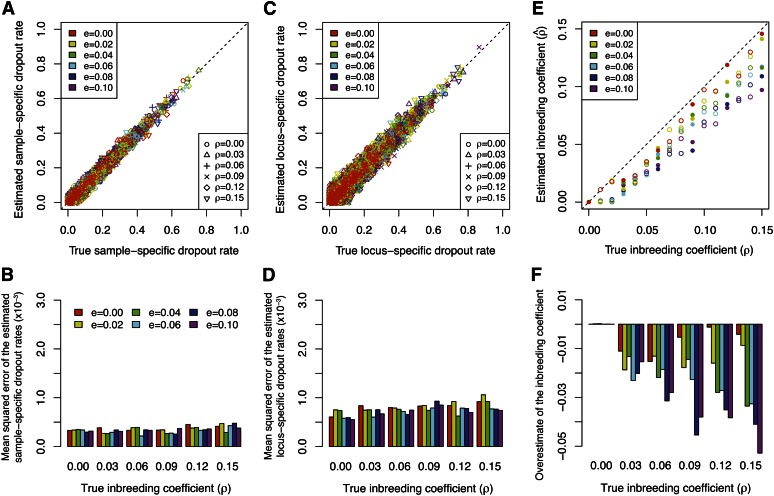
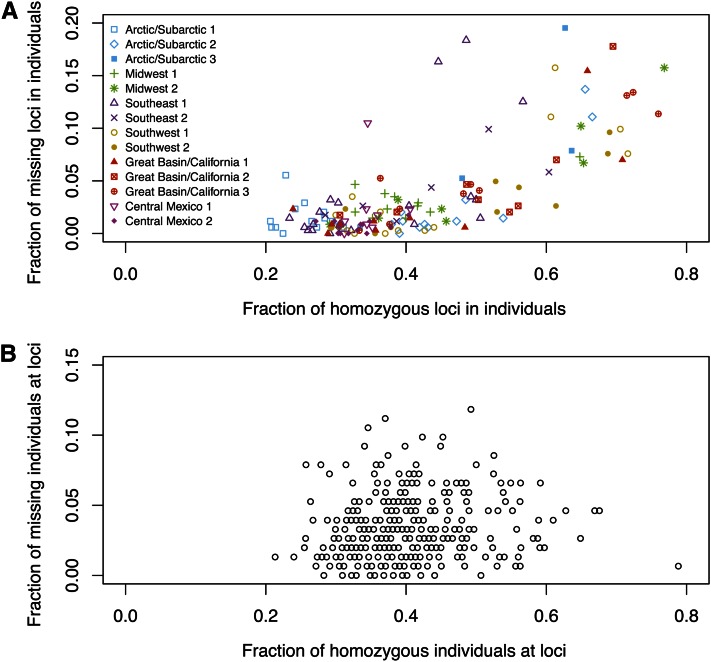
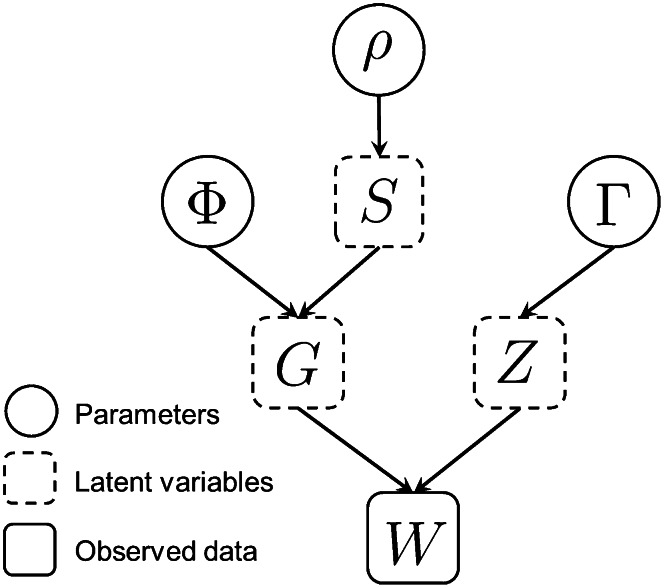
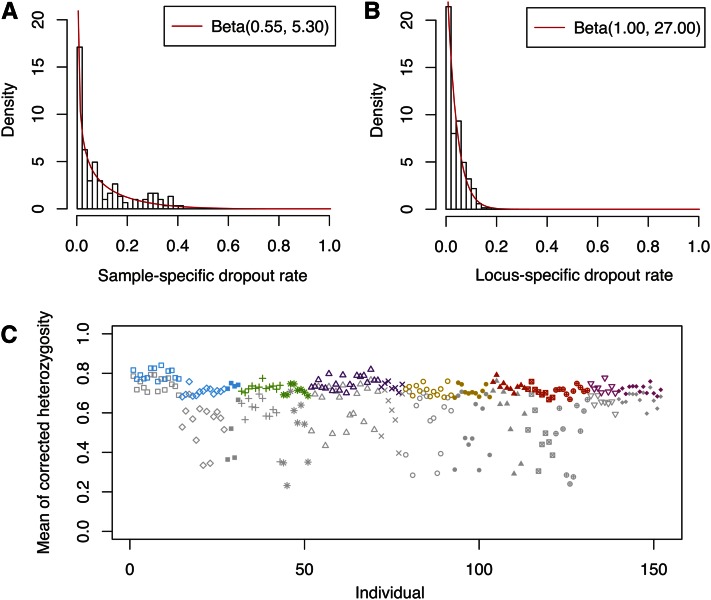
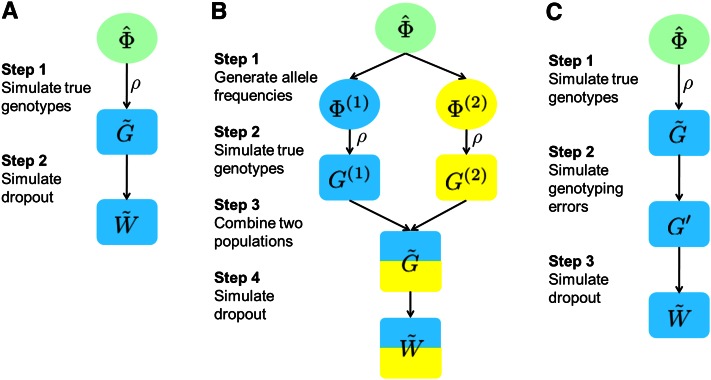
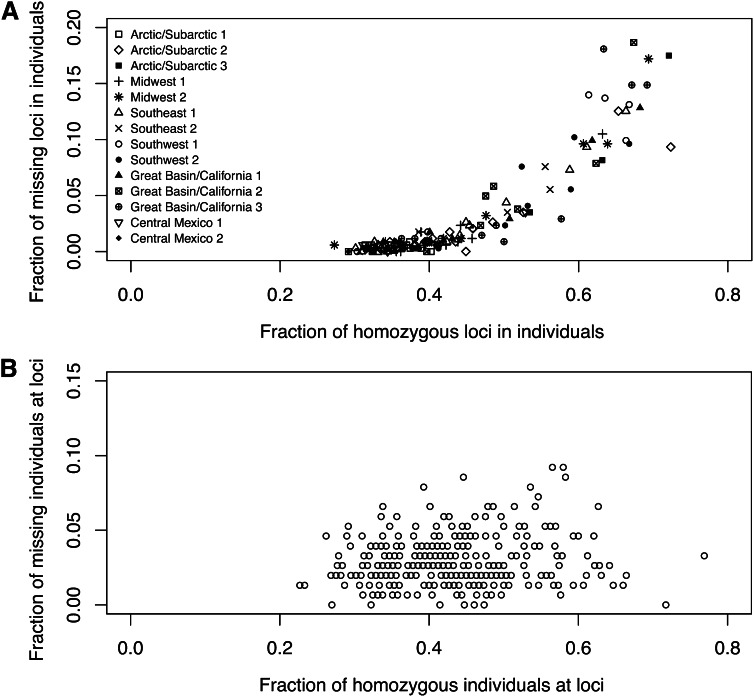
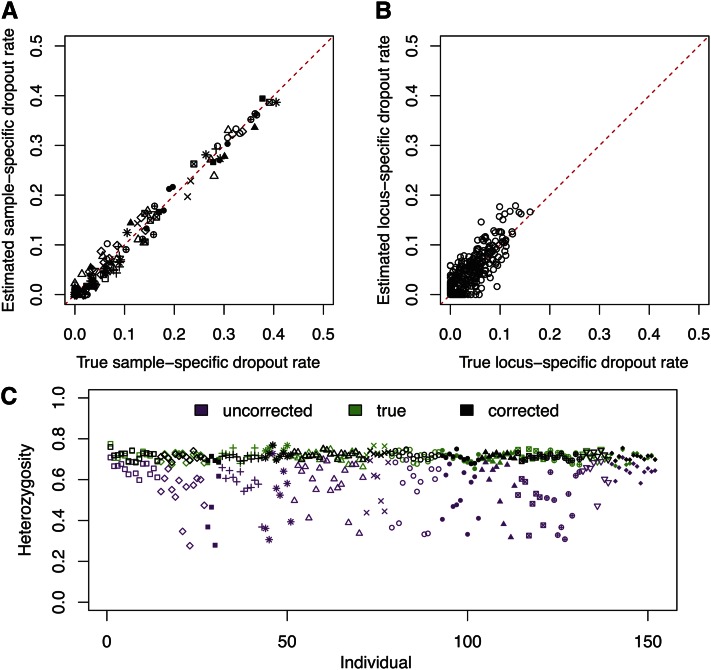
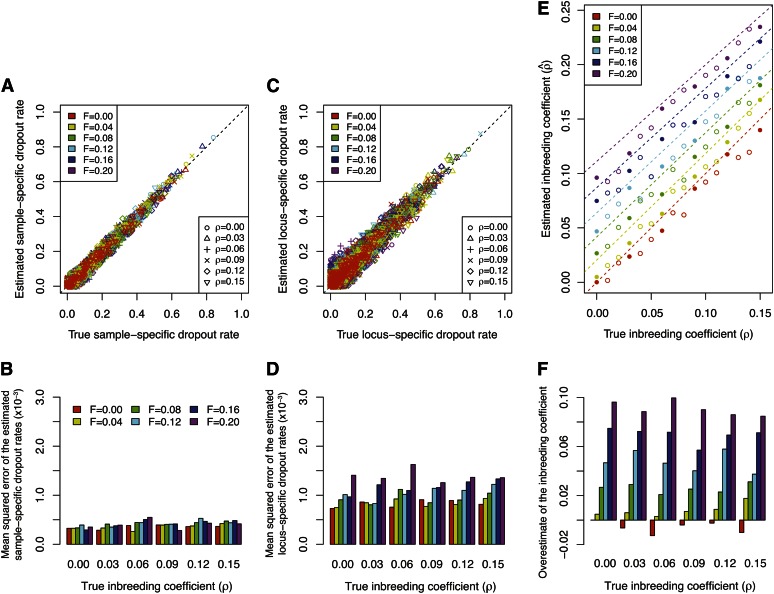
Allelic dropout is a commonly observed source of missing data in microsatellite genotypes, in which one or both allelic copies at a locus fail to be amplified by the polymerase chain reaction. Especially for samples with poor DNA quality, this problem causes a downward bias in estimates of observed heterozygosity and an upward bias in estimates of inbreeding, owing to mistaken classifications of heterozygotes as homozygotes when one of the two copies drops out. One general approach for avoiding allelic dropout involves repeated genotyping of homozygous loci to minimize the effects of experimental error. Existing computational alternatives often require replicate genotyping as well. These approaches, however, are costly and are suitable only when enough DNA is available for repeated genotyping. In this study, we propose a maximum-likelihood approach together with an expectation-maximization algorithm to jointly estimate allelic dropout rates and allele frequencies when only one set of nonreplicated genotypes is available. Our method considers estimates of allelic dropout caused by both sample-specific factors and locus-specific factors, and it allows for deviation from Hardy-Weinberg equilibrium owing to inbreeding. Using the estimated parameters, we correct the bias in the estimation of observed heterozygosity through the use of multiple imputations of alleles in cases where dropout might have occurred. With simulated data, we show that our method can (1) effectively reproduce patterns of missing data and heterozygosity observed in real data; (2) correctly estimate model parameters, including sample-specific dropout rates, locus-specific dropout rates, and the inbreeding coefficient; and (3) successfully correct the downward bias in estimating the observed heterozygosity. We find that our method is fairly robust to violations of model assumptions caused by population structure and by genotyping errors from sources other than allelic dropout. Because the data sets imputed under our model can be investigated in additional subsequent analyses, our method will be useful for preparing data for applications in diverse contexts in population genetics and molecular ecology.
等位基因缺失是微卫星基因型中常见的缺失数据来源,在这种情况下,一个或两个基因座的等位基因拷贝未能被聚合酶链反应扩增。对于 DNA 质量较差的样本尤其如此,由于一个拷贝缺失时,杂合子被错误地分类为纯合子,因此这种问题会导致观察到的杂合度估计值向下偏差,近交系数估计值向上偏差。避免等位基因缺失的一种通用方法是重复对纯合基因座进行基因分型,以最小化实验误差的影响。现有的计算替代方法通常也需要重复基因分型。然而,这些方法成本高昂,并且仅在有足够的 DNA 可用于重复基因分型时才适用。在这项研究中,我们提出了一种最大似然方法,并结合期望最大化算法,在仅提供一组非重复基因型的情况下联合估计等位基因缺失率和等位基因频率。我们的方法考虑了由样本特异性因素和基因座特异性因素引起的等位基因缺失估计值,并且允许由于近交而偏离哈迪-温伯格平衡。使用估计的参数,我们通过在可能发生缺失的情况下对等位基因进行多次插补,来纠正观察到的杂合度估计值的偏差。使用模拟数据,我们表明我们的方法可以(1)有效地再现实际数据中观察到的缺失数据和杂合度模式;(2)正确估计模型参数,包括样本特异性缺失率、基因座特异性缺失率和近交系数;(3)成功纠正估计观察到的杂合度的向下偏差。我们发现,我们的方法对于由群体结构和除等位基因缺失以外的其他来源的基因分型错误引起的模型假设违反具有相当的稳健性。由于我们的模型下推断的数据可以在其他后续分析中进行研究,因此我们的方法将有助于为群体遗传学和分子生态学中不同背景下的应用准备数据。