Department of Molecular and Cellular Physiology, Beckman Center, Stanford University, Palo Alto, California, USA.
PLoS Comput Biol. 2012;8(8):e1002671. doi: 10.1371/journal.pcbi.1002671. Epub 2012 Aug 30.
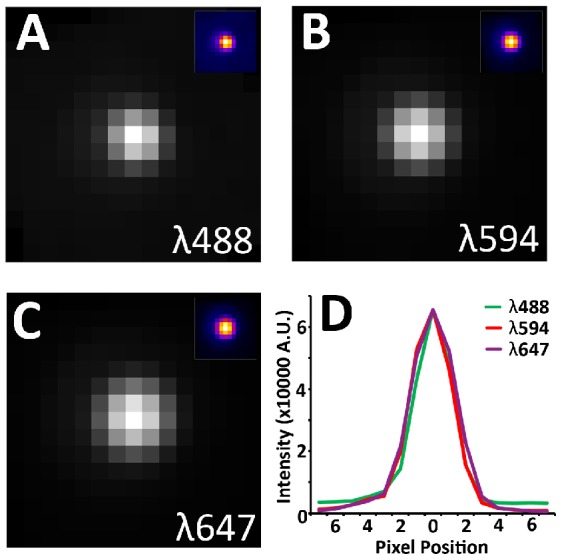
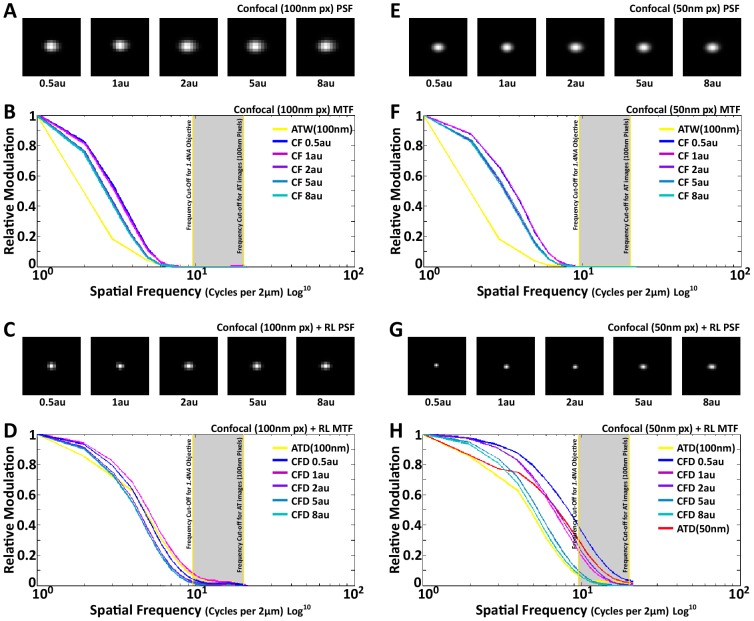
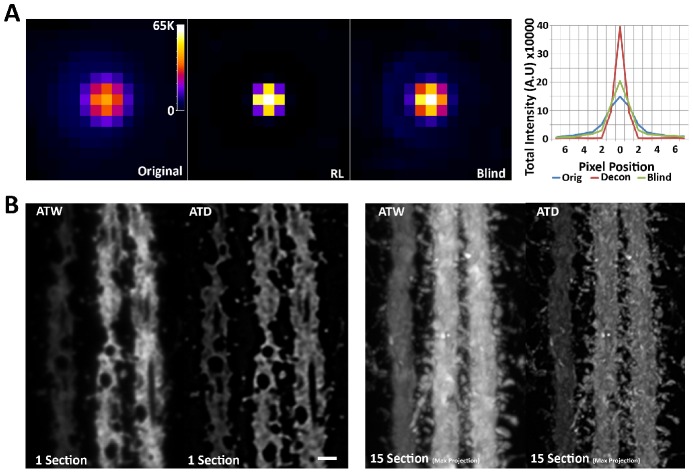
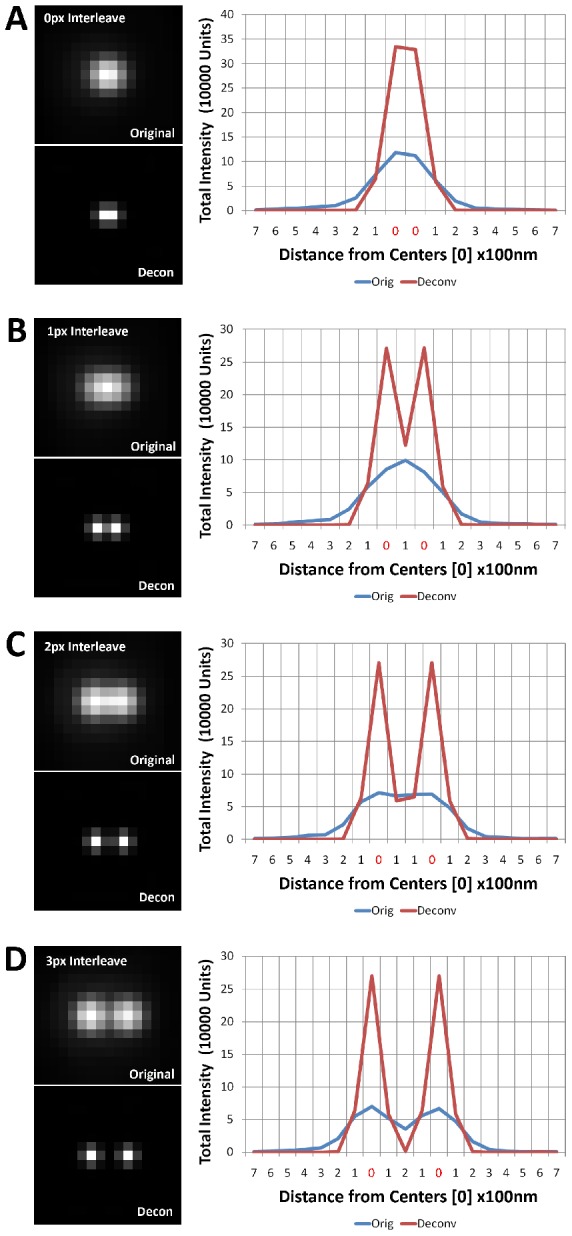
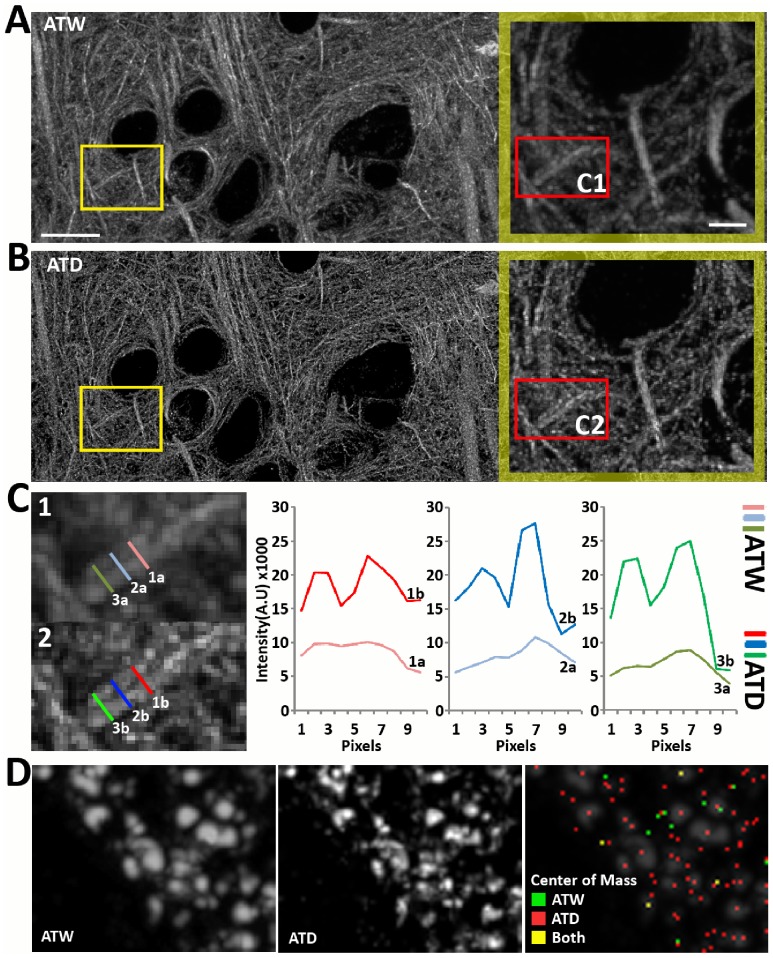
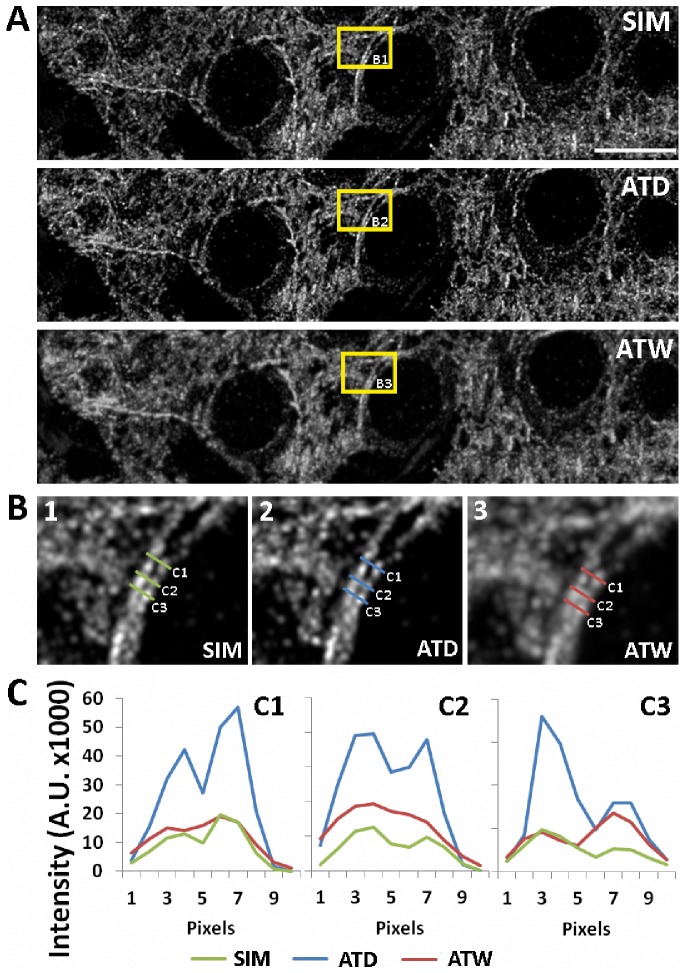
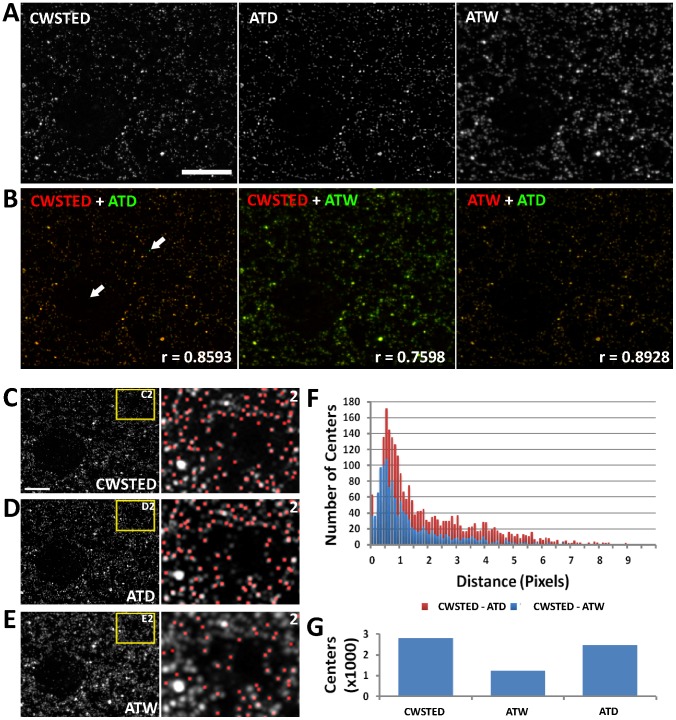
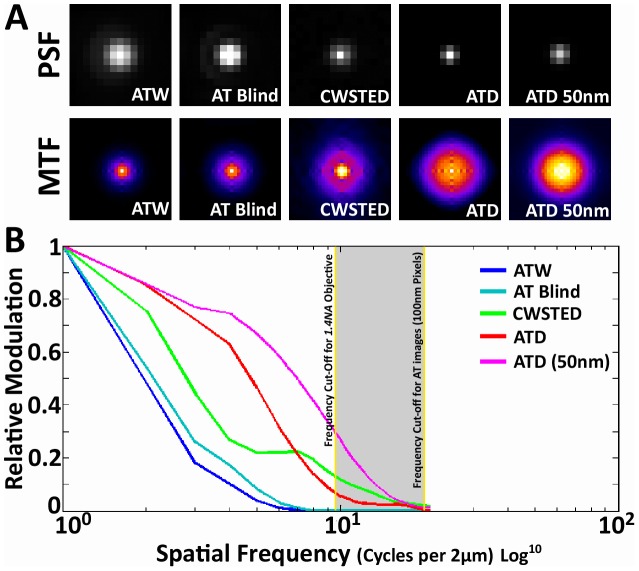
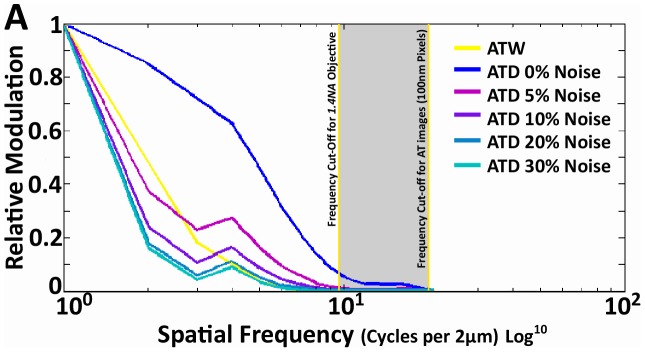
Photon diffraction limits the resolution of conventional light microscopy at the lateral focal plane to 0.61λ/NA (λ = wavelength of light, NA = numerical aperture of the objective) and at the axial plane to 1.4nλ/NA(2) (n = refractive index of the imaging medium, 1.51 for oil immersion), which with visible wavelengths and a 1.4NA oil immersion objective is -220 nm and -600 nm in the lateral plane and axial plane respectively. This volumetric resolution is too large for the proper localization of protein clustering in subcellular structures. Here we combine the newly developed proteomic imaging technique, Array Tomography (AT), with its native 50-100 nm axial resolution achieved by physical sectioning of resin embedded tissue, and a 2D maximum likelihood deconvolution method, based on Bayes' rule, which significantly improves the resolution of protein puncta in the lateral plane to allow accurate and fast computational segmentation and analysis of labeled proteins. The physical sectioning of AT allows tissue specimens to be imaged at the physical optimum of modern high NA plan-apochormatic objectives. This translates to images that have little out of focus light, minimal aberrations and wave-front distortions. Thus, AT is able to provide images with truly invariant point spread functions (PSF), a property critical for accurate deconvolution. We show that AT with deconvolution increases the volumetric analytical fidelity of protein localization by significantly improving the modulation of high spatial frequencies up to and potentially beyond the spatial frequency cut-off of the objective. Moreover, we are able to achieve this improvement with no noticeable introduction of noise or artifacts and arrive at object segmentation and localization accuracies on par with image volumes captured using commercial implementations of super-resolution microscopes.
光子衍射将传统的明场显微镜在横向焦平面的分辨率限制在 0.61λ/NA(λ=光的波长,NA=物镜的数值孔径),在轴向平面的分辨率限制在 1.4nλ/NA(2)(n=成像介质的折射率,油浸时为 1.51),对于可见波长和 1.4NA 油浸物镜,在横向平面和轴向平面的分辨率分别为-220nm 和-600nm。这种体积分辨率对于亚细胞结构中蛋白质聚簇的正确定位来说太大了。在这里,我们将新开发的蛋白质组学成像技术——阵列断层成像(AT)与它通过对树脂包埋组织进行物理切片获得的 50-100nm 的固有轴向分辨率相结合,以及一种基于贝叶斯规则的 2D 最大似然反卷积方法,该方法显著提高了蛋白质斑点在横向平面上的分辨率,从而能够准确快速地对标记蛋白进行计算分割和分析。AT 的物理切片允许对组织标本在现代高 NA 平面消色差物镜的物理最佳状态下进行成像。这转化为具有很少离焦光、最小像差和波前变形的图像。因此,AT 能够提供具有真正不变的点扩散函数(PSF)的图像,这是准确反卷积的关键属性。我们表明,通过反卷积,AT 增加了蛋白质定位的体积分析保真度,显著提高了高达并可能超过物镜空间频率截止的高空间频率的调制。此外,我们能够在不引入明显噪声或伪影的情况下实现这种改进,并达到与使用商业实施的超分辨率显微镜捕获的图像体积相当的对象分割和定位精度。