Department of Genome Sciences, University of Washington, Seattle, WA, USA.
BMC Bioinformatics. 2012 Nov 19;13:308. doi: 10.1186/1471-2105-13-308.
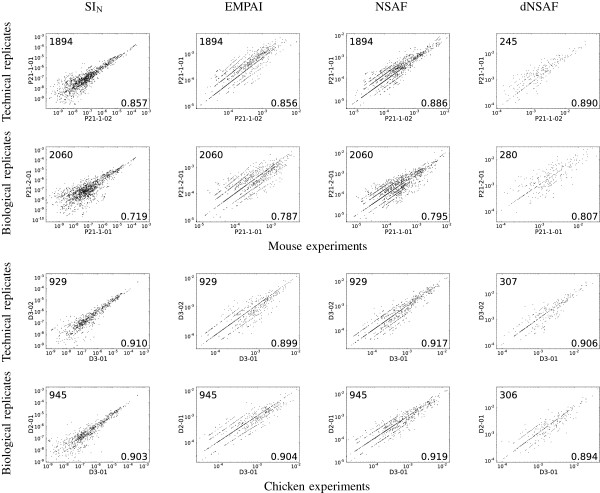
Spectral counting methods provide an easy means of identifying proteins with differing abundances between complex mixtures using shotgun proteomics data. The crux spectral-counts command, implemented as part of the Crux software toolkit, implements four previously reported spectral counting methods, the spectral index (SI(N)), the exponentially modified protein abundance index (emPAI), the normalized spectral abundance factor (NSAF), and the distributed normalized spectral abundance factor (dNSAF).
We compared the reproducibility and the linearity relative to each protein's abundance of the four spectral counting metrics. Our analysis suggests that NSAF yields the most reproducible counts across technical and biological replicates, and both SI(N) and NSAF achieve the best linearity.
With the crux spectral-counts command, Crux provides open-source modular methods to analyze mass spectrometry data for identifying and now quantifying peptides and proteins. The C++ source code, compiled binaries, spectra and sequence databases are available at http://noble.gs.washington.edu/proj/crux-spectral-counts.
谱计数方法为使用 shotgun 蛋白质组学数据在复杂混合物中识别具有不同丰度的蛋白质提供了一种简便的方法。作为 Crux 软件工具包的一部分实现的 crux spectral-counts 命令实现了四种先前报道的谱计数方法,即谱指数 (SI(N))、指数修正的蛋白质丰度指数 (emPAI)、归一化谱丰度因子 (NSAF) 和分布式归一化谱丰度因子 (dNSAF)。
我们比较了四种谱计数指标的重现性和相对于每种蛋白质丰度的线性关系。我们的分析表明,NSAF 在技术和生物学重复中产生最可重复的计数,而 SI(N) 和 NSAF 都能达到最佳的线性度。
使用 crux spectral-counts 命令,Crux 提供了用于分析质谱数据以识别和现在定量肽和蛋白质的开源模块化方法。C++源代码、编译二进制文件、光谱和序列数据库可在 http://noble.gs.washington.edu/proj/crux-spectral-counts 上获得。