Cembran Alessandro, Provorse Makenzie R, Wang Changwei, Wu Wei, Gao Jiali
Department of Chemistry, Digital Technology Center and Supercomputing Institute, University of Minnesota, Minneapolis, MN 55455.
J Chem Theory Comput. 2012 Nov 13;8(11):4347-4358. doi: 10.1021/ct3004595. Epub 2012 Sep 4.
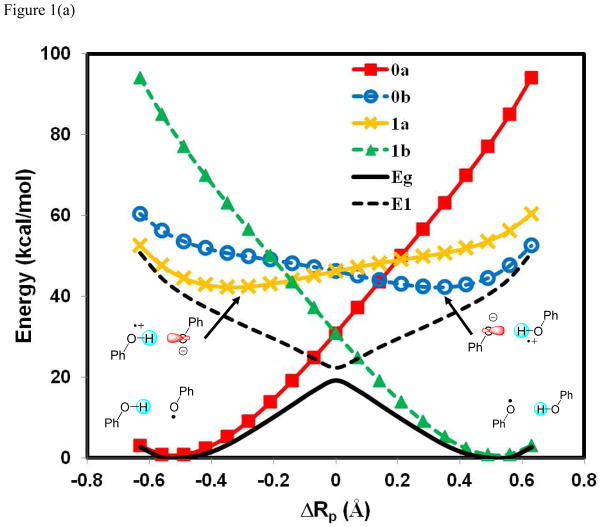
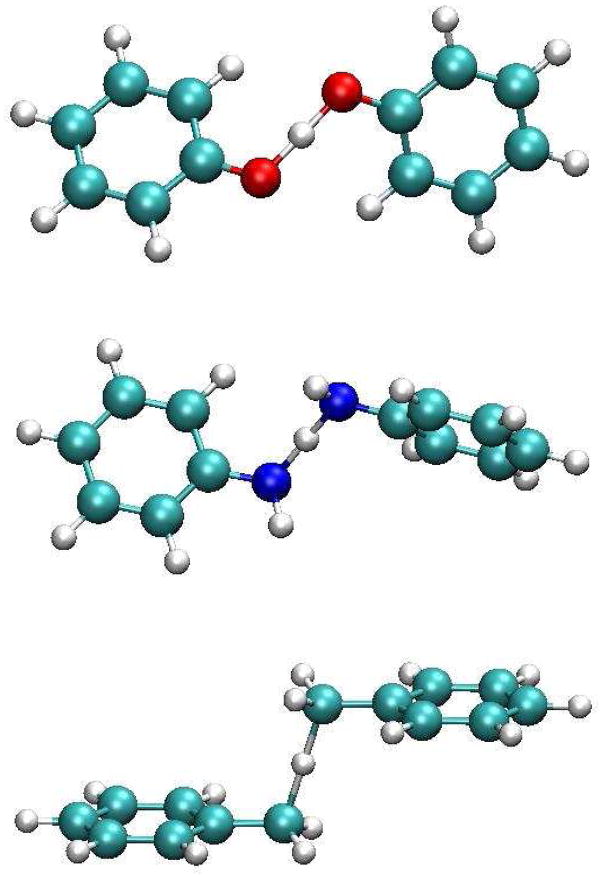
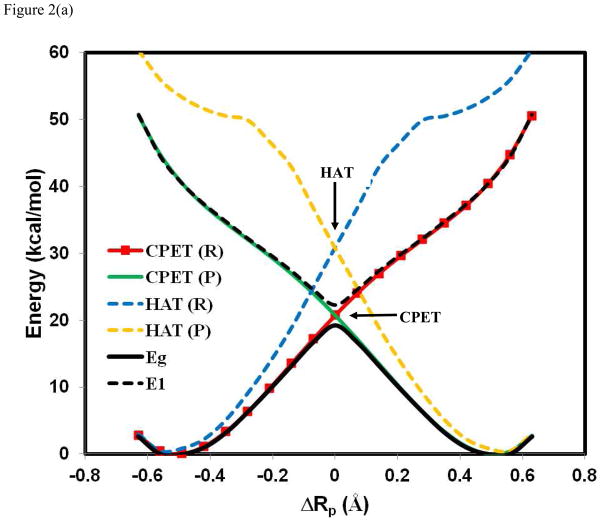
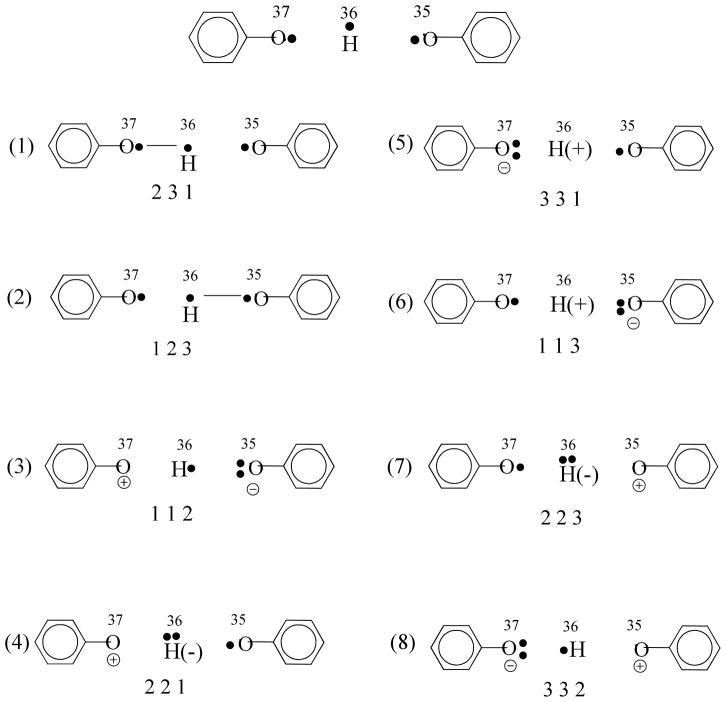
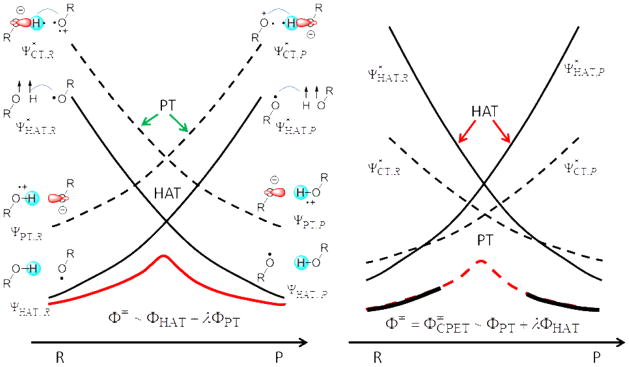
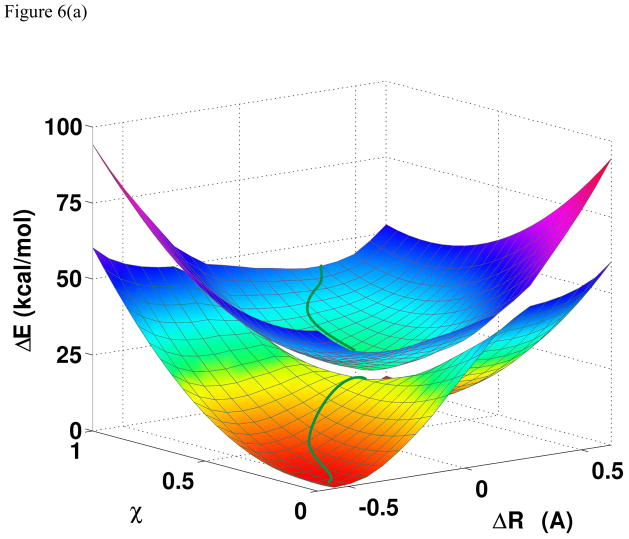
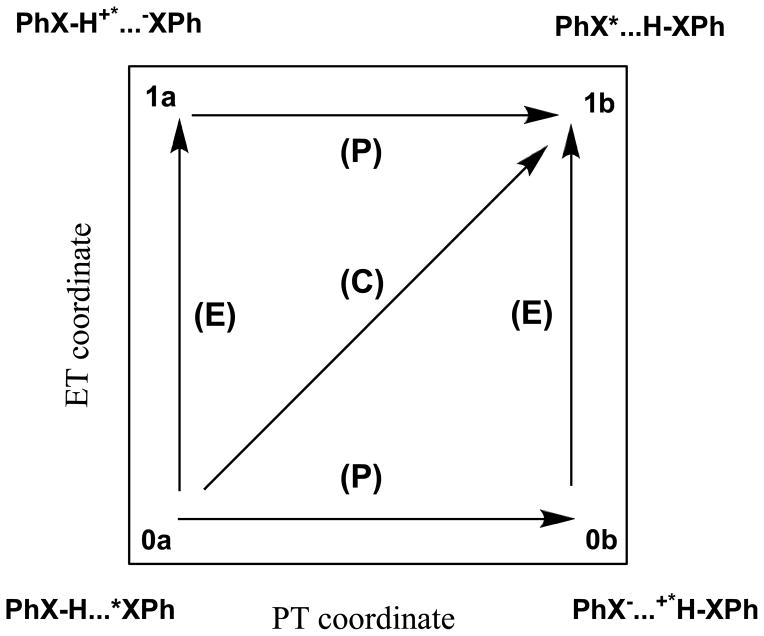
A critical element in theoretical characterization of the mechanism of proton-coupled electron transfer (PCET) reactions, including hydrogen atom transfer (HAT), is the formulation of the electron and proton localized diabatic states, based on which a More O'Ferrall-Jencks diagram can be represented to determine the step-wise and concerted nature of the reaction. Although the More O'Ferrall-Jencks diabatic states have often been used empirically to develop theoretical models for PCET reactions, the potential energy surfaces for these states have never been determined directly based on first principles calculations using electronic structure theory. The difficulty is due to a lack of practical method to constrain electron and proton localized diabatic states in wave function or density functional theory calculations. Employing a multistate density functional theory (MSDFT), in which the electron and proton localized diabatic configurations are constructed through block-localization of Kohn-Sham orbitals, we show that distinction between concerted proton-electron transfer (CPET) and HAT, which are not distinguishable experimentally from phenomenological kinetic data, can be made by examining the third dimension of a More O'Ferrall-Jencks diagram that includes both the ground and excited state potential surfaces. In addition, we formulate a pair of effective two-state valence bond models to represent the CPET and HAT mechanisms. We found that the lower energy of the CPET and HAT effective diabatic states at the intersection point can be used as an energetic criterion to distinguish the two mechanisms. In the isoelectronic series of hydrogen exchange reaction in (PhX)(2)H(•), where X = O, NH, and CH(2), there is a continuous transition from a CPET mechanism for the phenoxy radical-phenol pair to a HAT process for benzyl radical and toluene, while the reaction between PhNH(2) and PhNH(•) has a mechanism intermediate of CPET and HAT. The electronically nonadiabatic nature of the CPET mechanism in the phenol system can be attributed to the overlap interactions between the ground and excited state surfaces, resulting in roughly orthogonal minimum energy paths on the adiabatic ground and excited state potential energy surfaces. On the other hand, the minimum energy path on the adiabatic ground state for the HAT mechanism coincides with that on the excited state, producing a large electronic coupling that separates the two surfaces by more than 120 kcal/mol.
质子耦合电子转移(PCET)反应机制的理论表征中的一个关键要素,包括氢原子转移(HAT),是电子和质子局域非绝热态的公式化,基于此可以绘制一个莫尔·奥费拉尔 - 詹克斯(More O'Ferrall-Jencks)图来确定反应的分步和协同性质。尽管莫尔·奥费拉尔 - 詹克斯非绝热态常常被经验性地用于开发PCET反应的理论模型,但这些态的势能面从未基于使用电子结构理论的第一性原理计算直接确定。困难在于缺乏在波函数或密度泛函理论计算中约束电子和质子局域非绝热态的实用方法。采用多态密度泛函理论(MSDFT),其中电子和质子局域非绝热构型通过科恩 - 沈(Kohn-Sham)轨道的块定位构建,我们表明,通过检查包含基态和激发态势能面的莫尔·奥费拉尔 - 詹克斯图的第三维,可以区分协同质子 - 电子转移(CPET)和HAT,这两者从唯象动力学数据在实验上无法区分。此外,我们公式化了一对有效的双态价键模型来表示CPET和HAT机制。我们发现,在交点处CPET和HAT有效非绝热态的较低能量可以用作区分这两种机制的能量标准。在(PhX)₂H•中的氢交换反应的等电子系列中,其中X = O、NH和CH₂,从苯氧基自由基 - 苯酚对的CPET机制到苄基自由基和甲苯的HAT过程存在连续转变,而PhNH₂和PhNH•之间的反应具有CPET和HAT中间的机制。苯酚体系中CPET机制的电子非绝热性质可归因于基态和激发态表面之间的重叠相互作用,导致绝热基态和激发态势能面上大致正交的最小能量路径。另一方面,HAT机制在绝热基态上的最小能量路径与激发态上的重合,产生了超过120千卡/摩尔的大电子耦合,将两个表面分开。