Laboratory for Informatics and Data Mining, Department of Computer and Information Science, Fordham University, New York, NY 10023, USA.
BMC Genomics. 2012;13 Suppl 8(Suppl 8):S12. doi: 10.1186/1471-2164-13-S8-S12. Epub 2012 Dec 17.
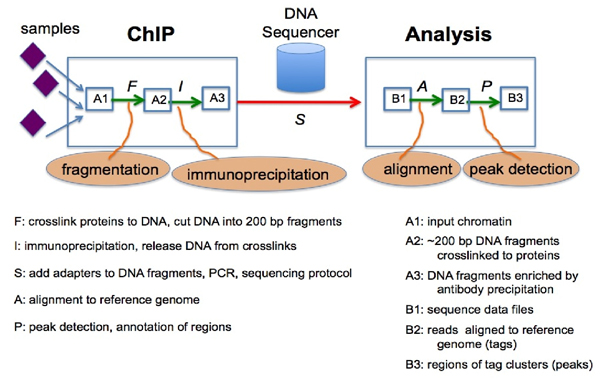
Due to the recent rapid development in ChIP-seq technologies, which uses high-throughput next-generation DNA sequencing to identify the targets of Chromatin Immunoprecipitation, there is an increasing amount of sequencing data being generated that provides us with greater opportunity to analyze genome-wide protein-DNA interactions. In particular, we are interested in evaluating and enhancing computational and statistical techniques for locating protein binding sites. Many peak detection systems have been developed; in this study, we utilize the following six: CisGenome, MACS, PeakSeq, QuEST, SISSRs, and TRLocator.
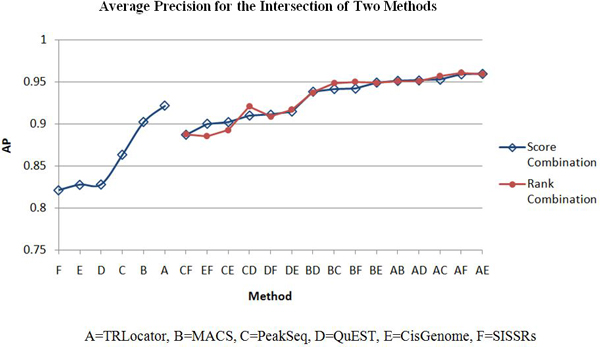
We define two methods to merge and rescore the regions of two peak detection systems and analyze the performance based on average precision and coverage of transcription start sites. The results indicate that ChIP-seq peak detection can be improved by fusion using score or rank combination.
Our method of combination and fusion analysis would provide a means for generic assessment of available technologies and systems and assist researchers in choosing an appropriate system (or fusion method) for analyzing ChIP-seq data. This analysis offers an alternate approach for increasing true positive rates, while decreasing false positive rates and hence improving the ChIP-seq peak identification process.
由于 ChIP-seq 技术的快速发展,该技术使用高通量的下一代 DNA 测序来鉴定染色质免疫沉淀的靶标,因此产生了越来越多的测序数据,这为我们提供了更多机会来分析全基因组蛋白-DNA 相互作用。特别是,我们有兴趣评估和增强用于定位蛋白质结合位点的计算和统计技术。已经开发了许多峰检测系统;在这项研究中,我们利用以下六个系统: CisGenome、MACS、PeakSeq、QuEST、SISSRs 和 TRLocator。
我们定义了两种合并和重新评分两种峰检测系统区域的方法,并基于转录起始位点的平均精度和覆盖度来分析性能。结果表明,通过使用分数或等级组合进行融合,可以提高 ChIP-seq 峰检测的性能。
我们的组合和融合分析方法可以为评估现有技术和系统提供一种通用的方法,并帮助研究人员选择适当的系统(或融合方法)来分析 ChIP-seq 数据。这种分析为提高真阳性率、降低假阳性率并因此改进 ChIP-seq 峰识别过程提供了一种替代方法。