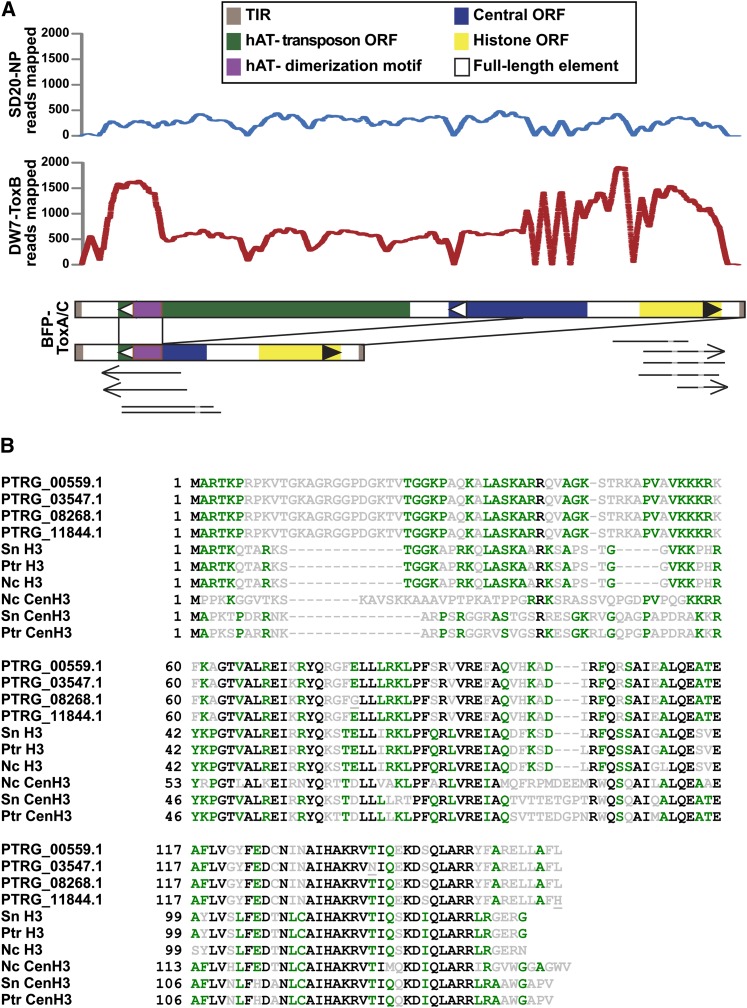
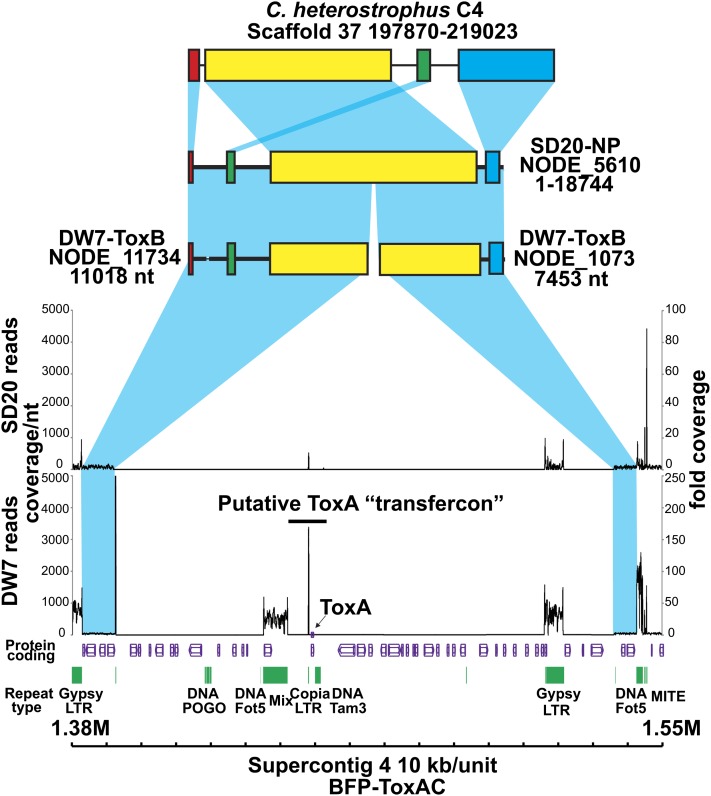
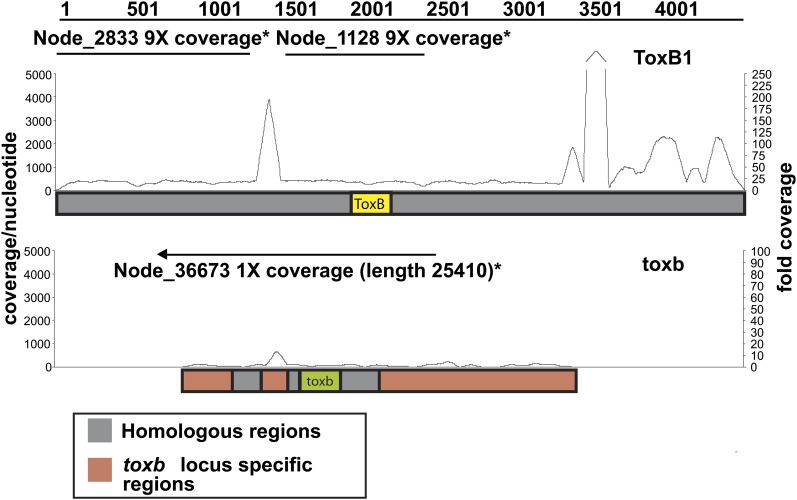
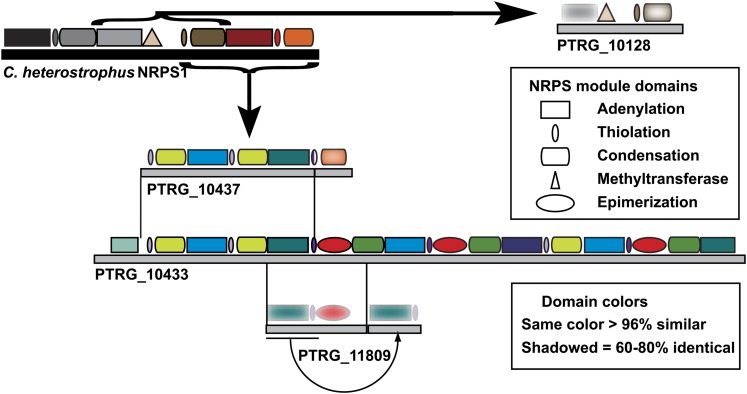
Department of Botany and Plant Pathology, Oregon State University, Corvallis, Oregon 97331-8530, USA.
G3 (Bethesda). 2013 Jan;3(1):41-63. doi: 10.1534/g3.112.004044. Epub 2013 Jan 1.
Pyrenophora tritici-repentis is a necrotrophic fungus causal to the disease tan spot of wheat, whose contribution to crop loss has increased significantly during the last few decades. Pathogenicity by this fungus is attributed to the production of host-selective toxins (HST), which are recognized by their host in a genotype-specific manner. To better understand the mechanisms that have led to the increase in disease incidence related to this pathogen, we sequenced the genomes of three P. tritici-repentis isolates. A pathogenic isolate that produces two known HSTs was used to assemble a reference nuclear genome of approximately 40 Mb composed of 11 chromosomes that encode 12,141 predicted genes. Comparison of the reference genome with those of a pathogenic isolate that produces a third HST, and a nonpathogenic isolate, showed the nonpathogen genome to be more diverged than those of the two pathogens. Examination of gene-coding regions has provided candidate pathogen-specific proteins and revealed gene families that may play a role in a necrotrophic lifestyle. Analysis of transposable elements suggests that their presence in the genome of pathogenic isolates contributes to the creation of novel genes, effector diversification, possible horizontal gene transfer events, identified copy number variation, and the first example of transduplication by DNA transposable elements in fungi. Overall, comparative analysis of these genomes provides evidence that pathogenicity in this species arose through an influx of transposable elements, which created a genetically flexible landscape that can easily respond to environmental changes.
禾谷核腔菌是一种引起小麦赤霉病的坏死型真菌,其对作物损失的贡献在过去几十年中显著增加。该真菌的致病性归因于其产生的宿主选择性毒素(HST),这些毒素以特定于宿主的方式被其宿主识别。为了更好地理解导致与该病原体相关的疾病发病率增加的机制,我们对三个禾谷核腔菌分离株的基因组进行了测序。一个产生两种已知 HST 的致病分离株被用于组装一个约 40 Mb 的参考核基因组,该基因组由 11 条染色体组成,编码 12,141 个预测基因。将参考基因组与产生第三种 HST 的一个致病分离株和一个非致病分离株的基因组进行比较,结果表明非致病分离株的基因组比两个病原体的基因组分化程度更高。对编码区的检查提供了候选的病原体特异性蛋白,并揭示了可能在坏死型生活方式中发挥作用的基因家族。转座元件的分析表明,它们在致病分离株基因组中的存在有助于新基因的产生、效应子多样化、可能的水平基因转移事件、鉴定拷贝数变异以及真菌中转座元件的转导复制的第一个例子。总的来说,这些基因组的比较分析提供了证据表明,该物种的致病性是通过转座元件的涌入而产生的,这创造了一个遗传上灵活的景观,可以轻松应对环境变化。