Molecular Pharmacology for Diabetes Group, Health Innovations Research Institute and School of Health Sciences, RMIT University, Melbourne, Victoria, Australia.
Diabetes. 2013 Jun;62(6):2095-105. doi: 10.2337/db12-1397. Epub 2013 Jan 24.
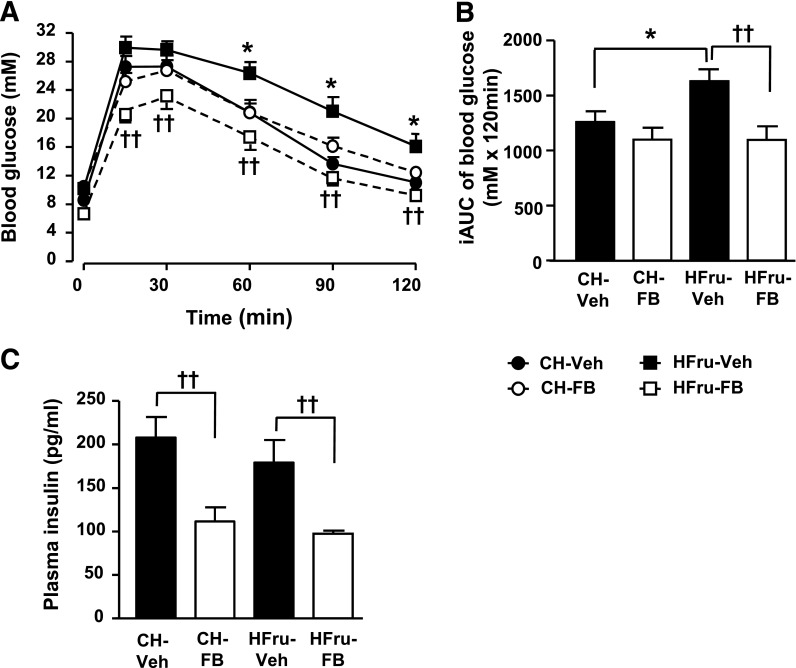
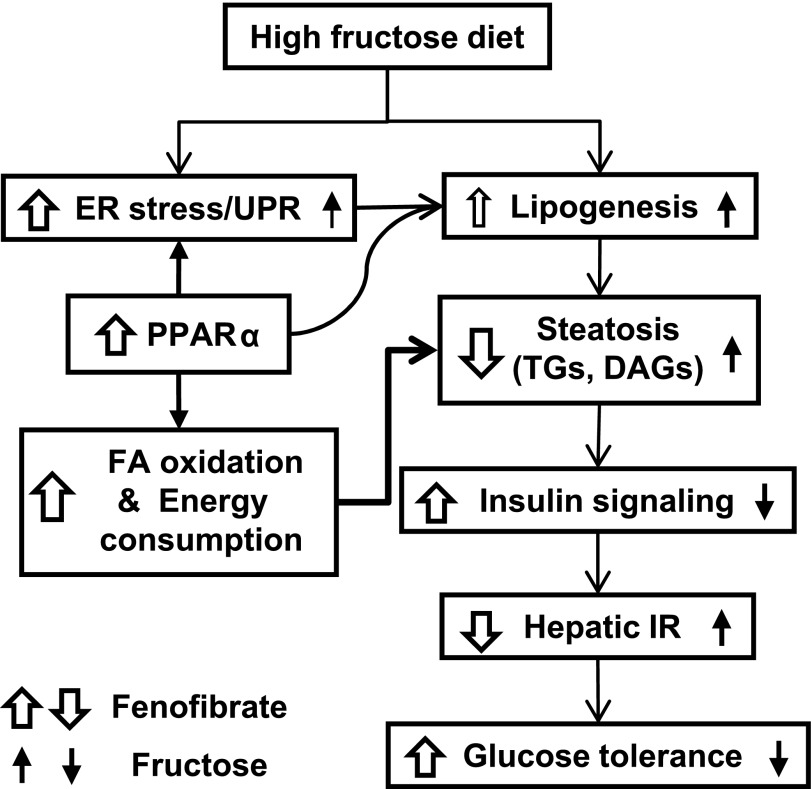
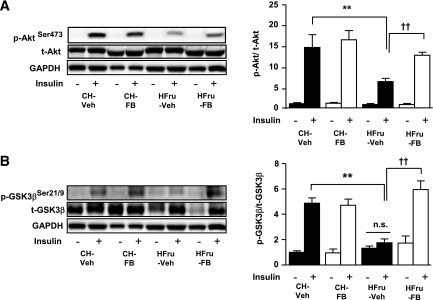
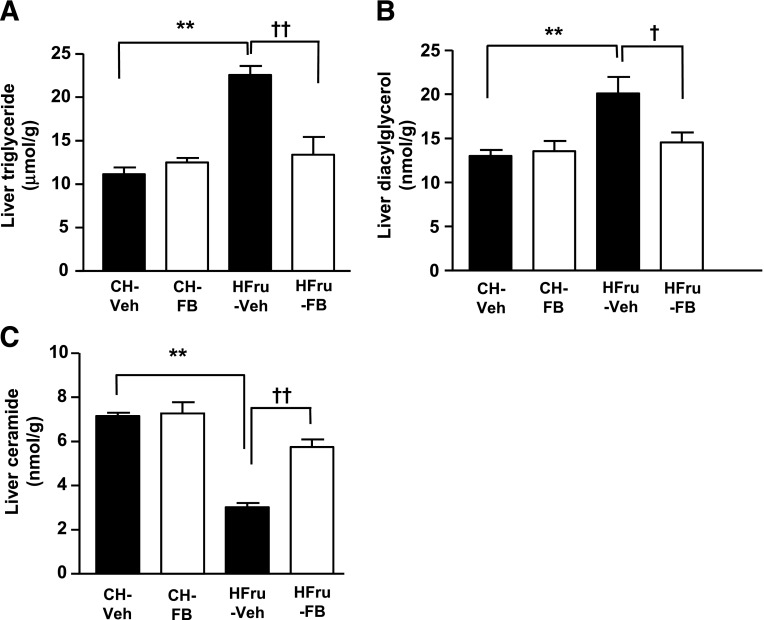
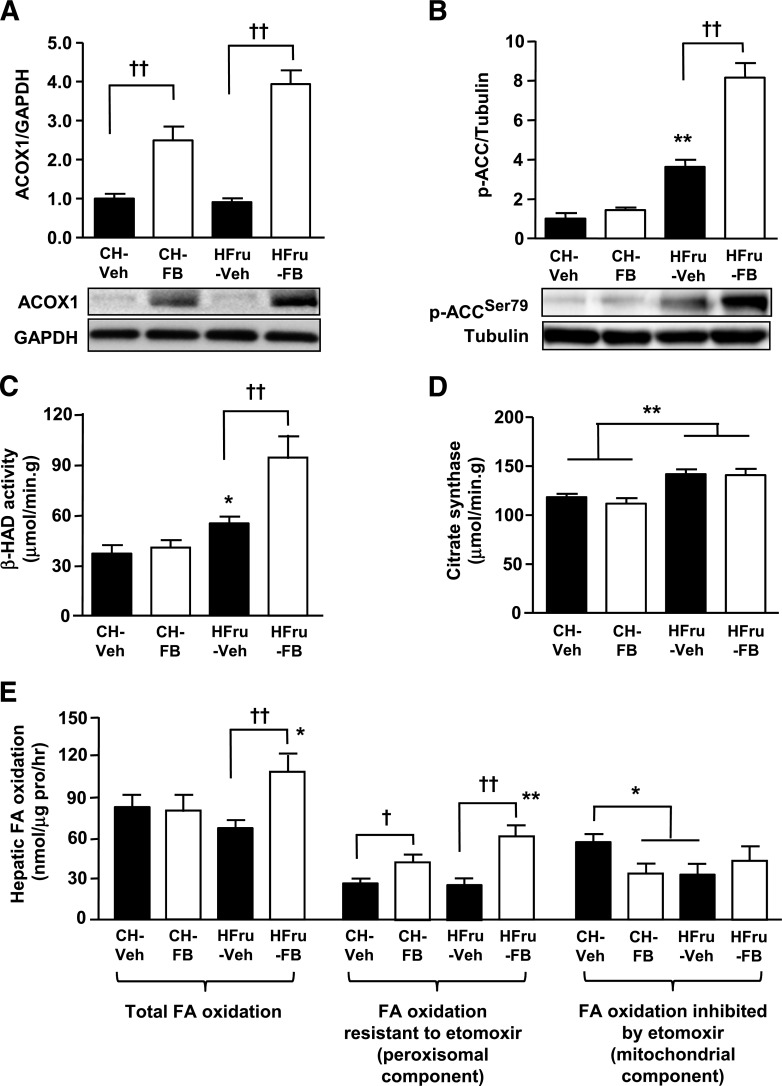
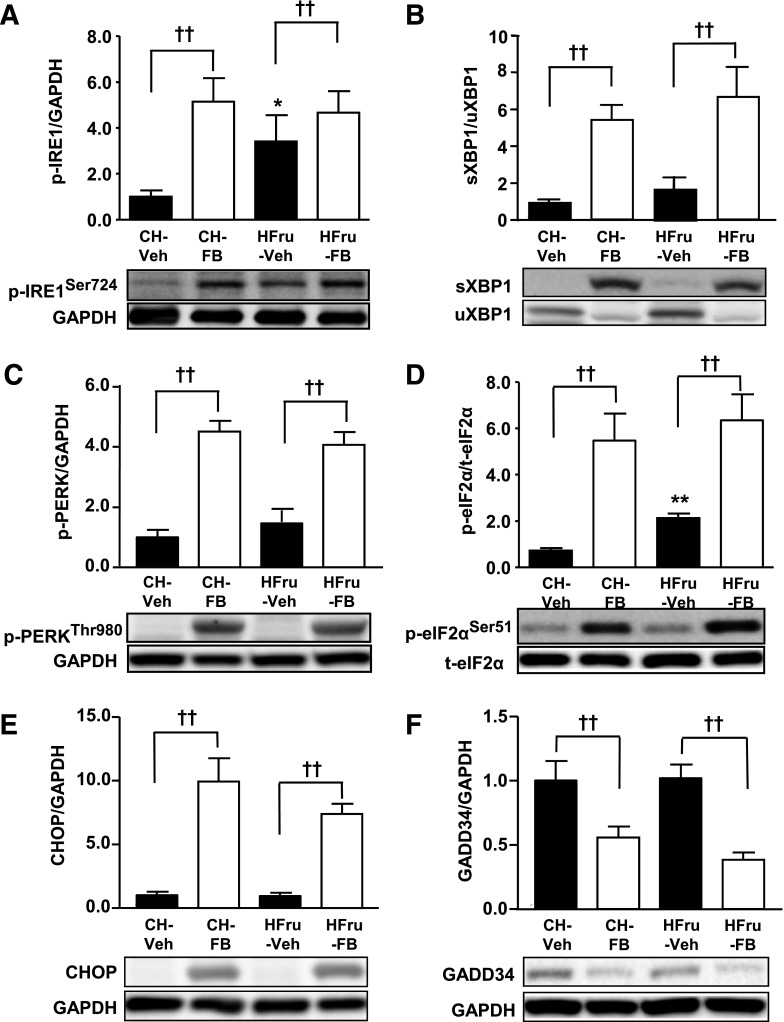
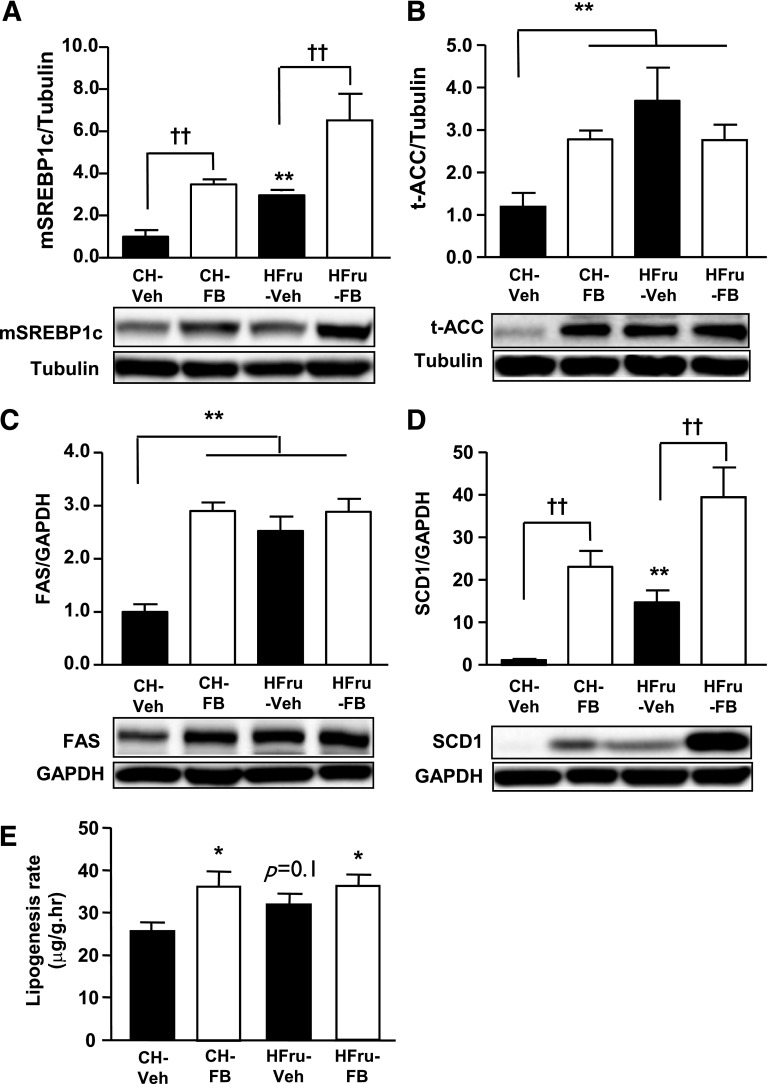
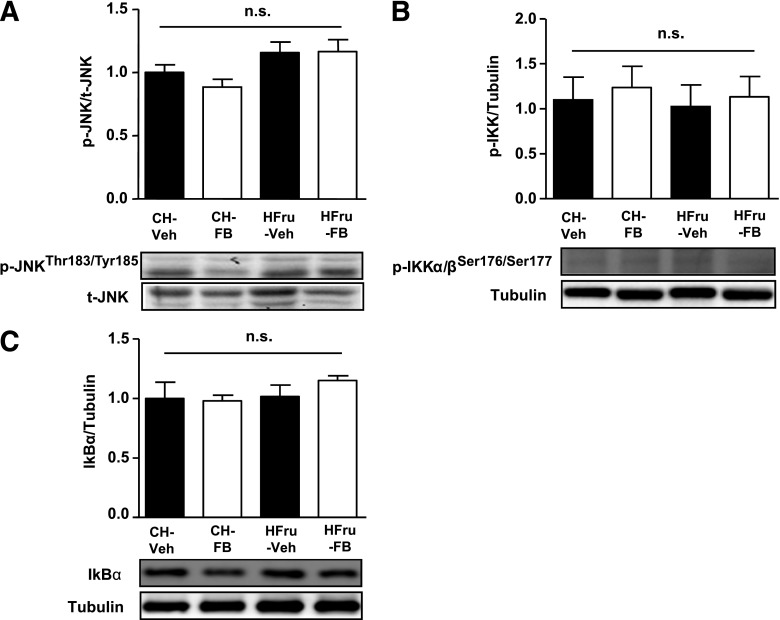
Endoplasmic reticulum (ER) stress is suggested to cause hepatic insulin resistance by increasing de novo lipogenesis (DNL) and directly interfering with insulin signaling through the activation of the c-Jun N-terminal kinase (JNK) and IκB kinase (IKK) pathway. The current study interrogated these two proposed mechanisms in a mouse model of hepatic insulin resistance induced by a high fructose (HFru) diet with the treatment of fenofibrate (FB) 100 mg/kg/day, a peroxisome proliferator-activated receptor α (PPARα) agonist known to reduce lipid accumulation while maintaining elevated DNL in the liver. FB administration completely corrected HFru-induced glucose intolerance, hepatic steatosis, and the impaired hepatic insulin signaling (pAkt and pGSK3β). Of note, both the IRE1/XBP1 and PERK/eIF2α arms of unfolded protein response (UPR) signaling were activated. While retaining the elevated DNL (indicated by the upregulation of SREBP1c, ACC, FAS, and SCD1 and [3H]H2O incorporation into lipids), FB treatment markedly increased fatty acid oxidation (indicated by induction of ACOX1, p-ACC, β-HAD activity, and [14C]palmitate oxidation) and eliminated the accumulation of diacylglycerols (DAGs), which is known to have an impact on insulin signaling. Despite the marked activation of UPR signaling, neither JNK nor IKK appeared to be activated. These findings suggest that lipid accumulation (mainly DAGs), rather than the activation of JNK or IKK, is pivotal for ER stress to cause hepatic insulin resistance. Therefore, by reducing the accumulation of deleterious lipids, activation of PPARα can ameliorate hepatic insulin resistance against increased ER stress.
内质网(ER)应激被认为通过增加从头脂肪生成(DNL)并通过激活 c-Jun N 端激酶(JNK)和 IκB 激酶(IKK)途径直接干扰胰岛素信号来导致肝胰岛素抵抗。本研究通过高果糖(HFru)饮食诱导的肝胰岛素抵抗小鼠模型研究了这两种拟议的机制,并用 100mg/kg/天的非诺贝特(FB)治疗,这是一种过氧化物酶体增殖物激活受体α(PPARα)激动剂,已知可减少脂质积累,同时保持肝脏中升高的 DNL。FB 给药完全纠正了 HFru 诱导的葡萄糖不耐受、肝脂肪变性和受损的肝胰岛素信号(pAkt 和 pGSK3β)。值得注意的是,未折叠蛋白反应(UPR)信号的 IRE1 / XBP1 和 PERK / eIF2α 臂均被激活。虽然保留了升高的 DNL(通过上调 SREBP1c、ACC、FAS 和 SCD1 以及 [3H]H2O 掺入脂质来指示),但 FB 处理显著增加了脂肪酸氧化(通过诱导 ACOX1、p-ACC、β-HAD 活性和 [14C]棕榈酸氧化来指示)并消除了二酰基甘油(DAG)的积累,已知 DAG 对胰岛素信号有影响。尽管 UPR 信号明显激活,但 JNK 或 IKK 似乎均未被激活。这些发现表明,脂质积累(主要是 DAG),而不是 JNK 或 IKK 的激活,对于 ER 应激导致肝胰岛素抵抗至关重要。因此,通过减少有害脂质的积累,激活 PPARα 可以改善肝胰岛素抵抗,减轻 ER 应激的增加。