Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden.
PLoS One. 2013;8(1):e53822. doi: 10.1371/journal.pone.0053822. Epub 2013 Jan 18.
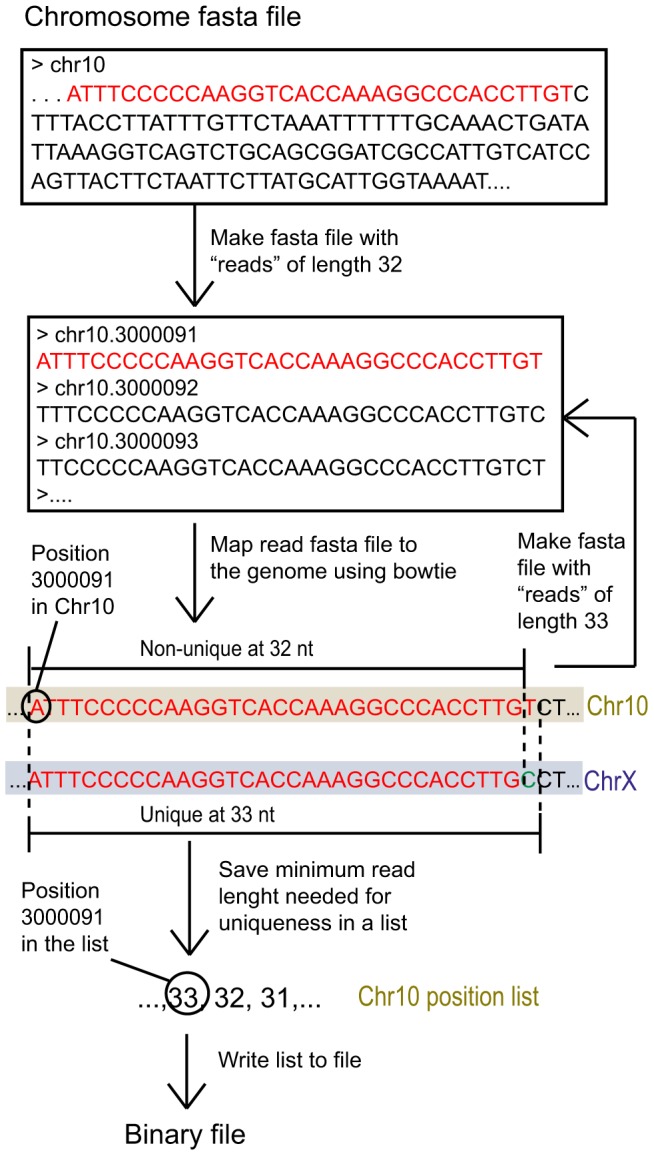
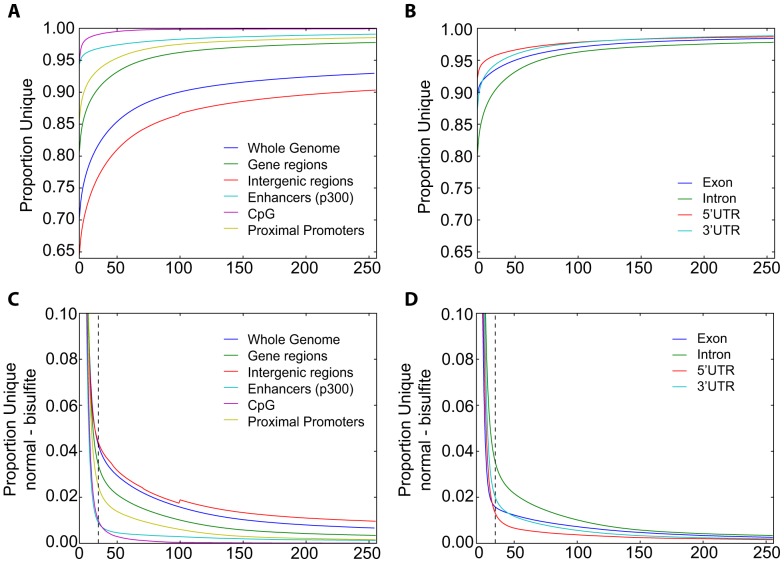
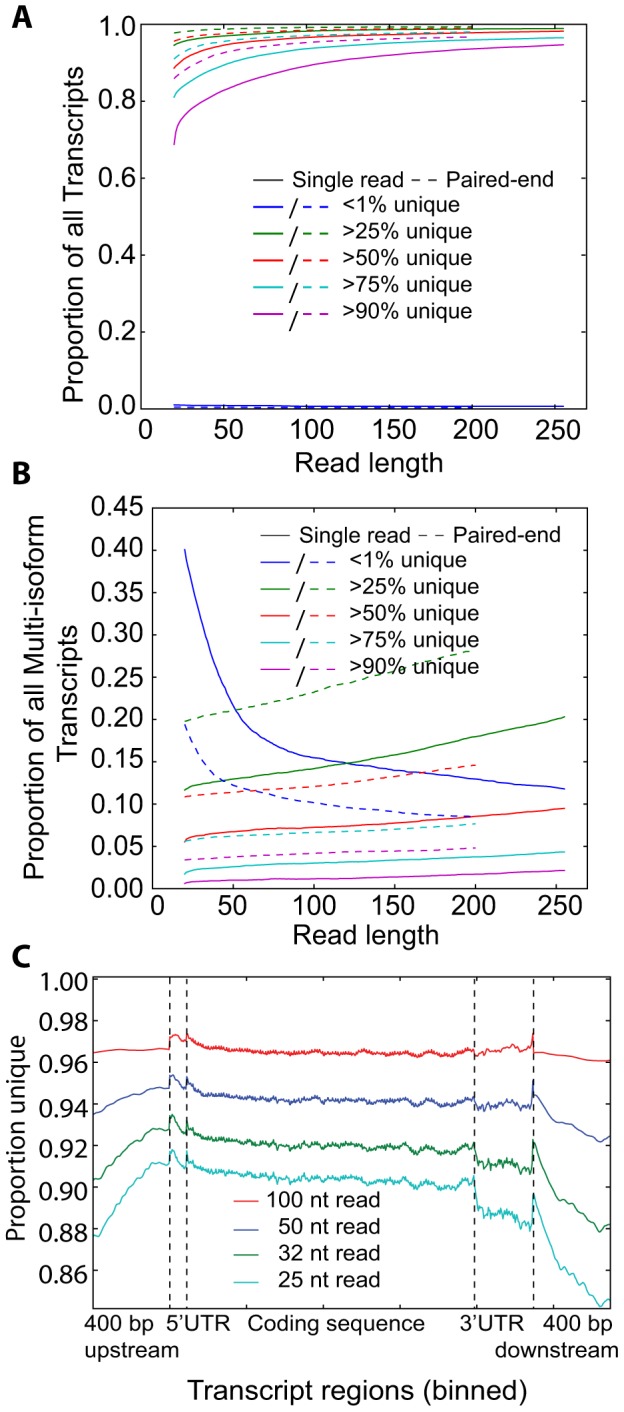
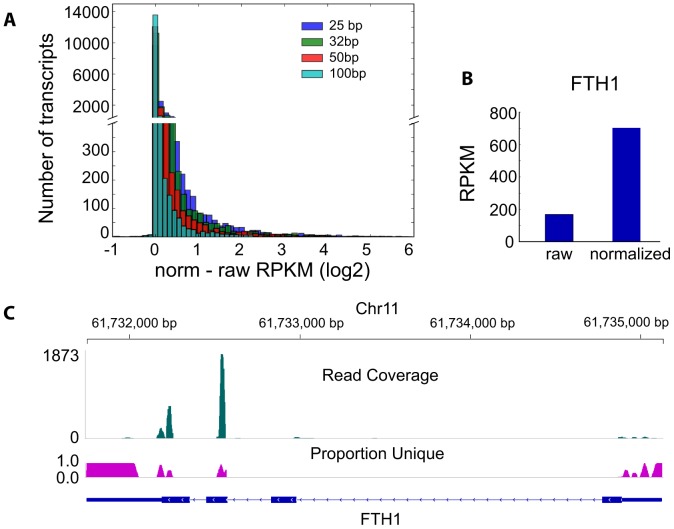
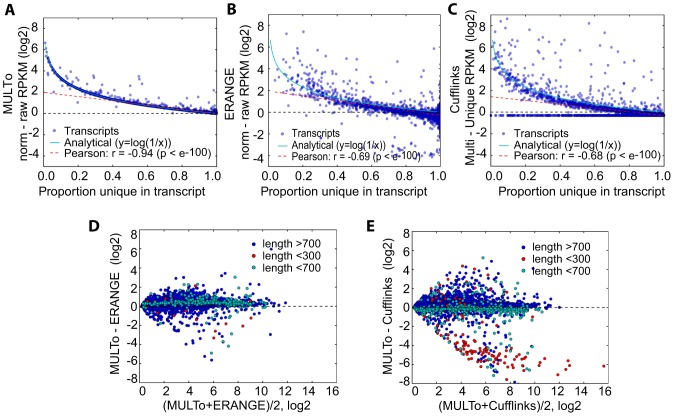
As next generation sequencing technologies are getting more efficient and less expensive, RNA-Seq is becoming a widely used technique for transcriptome studies. Computational analysis of RNA-Seq data often starts with the mapping of millions of short reads back to the genome or transcriptome, a process in which some reads are found to map equally well to multiple genomic locations (multimapping reads). We have developed the Minimum Unique Length Tool (MULTo), a framework for efficient and comprehensive representation of mappability information, through identification of the shortest possible length required for each genomic coordinate to become unique in the genome and transcriptome. Using the minimum unique length information, we have compared different uniqueness compensation approaches for transcript expression level quantification and demonstrate that the best compensation is achieved by discarding multimapping reads and correctly adjusting gene model lengths. We have also explored uniqueness within specific regions of the mouse genome and enhancer mapping experiments. Finally, by making MULTo available to the community we hope to facilitate the use of uniqueness compensation in RNA-Seq analysis and to eliminate the need to make additional mappability files.
随着下一代测序技术变得越来越高效和廉价,RNA-Seq 正成为转录组研究中广泛使用的技术。RNA-Seq 数据的计算分析通常从将数百万个短读段映射回基因组或转录组开始,在此过程中,一些读段被发现可以同样好地映射到多个基因组位置(多重映射读段)。我们通过确定每个基因组坐标在基因组和转录组中变得唯一所需的最短可能长度,开发了最小唯一长度工具(MULTo),这是一种高效和全面表示可映射性信息的框架。利用最小唯一长度信息,我们比较了不同的唯一性补偿方法来定量转录表达水平,并证明通过丢弃多重映射读段并正确调整基因模型长度可以实现最佳补偿。我们还探索了小鼠基因组特定区域和增强子映射实验中的独特性。最后,通过向社区提供 MULTo,我们希望促进在 RNA-Seq 分析中使用唯一性补偿,并消除制作额外可映射性文件的需求。