Clinical Pharmacology & Digestive Pathophysiology Unit, Department of Clinical & Experimental Medicine, University of Parma, Italy.
Curr Med Chem. 2013;20(19):2415-37. doi: 10.2174/09298673113209990115.
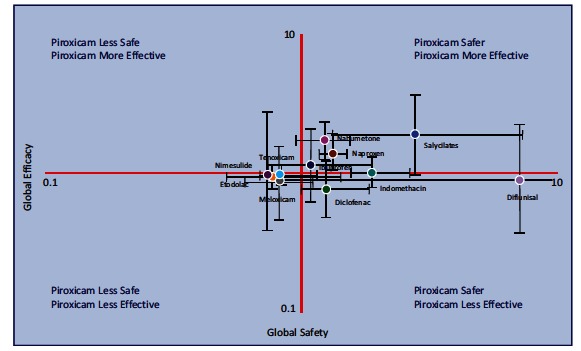
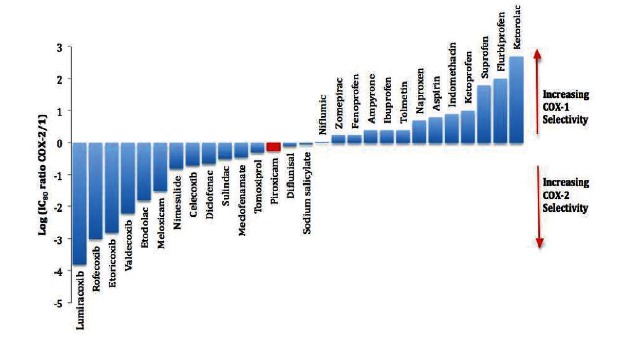
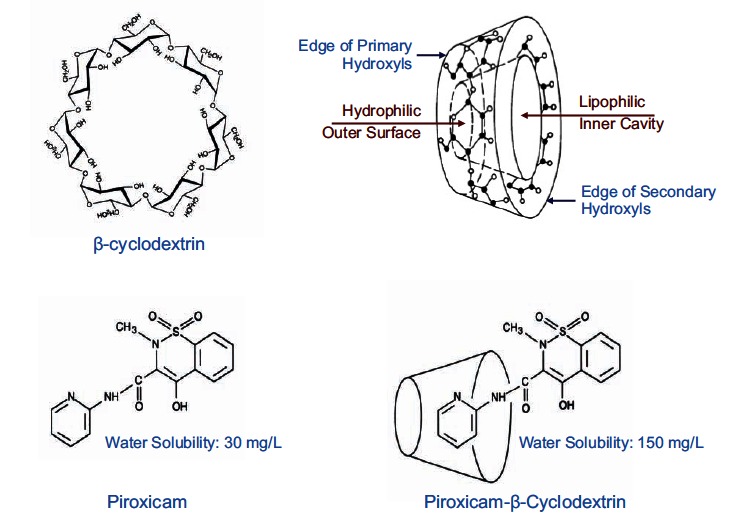
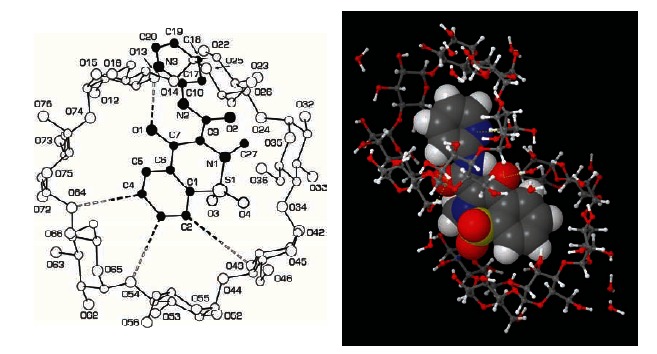
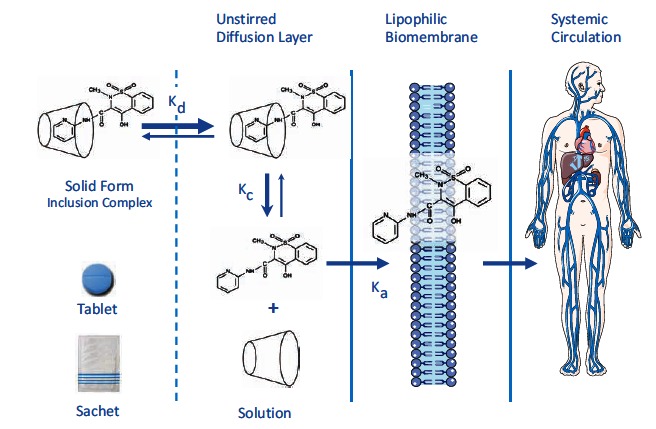
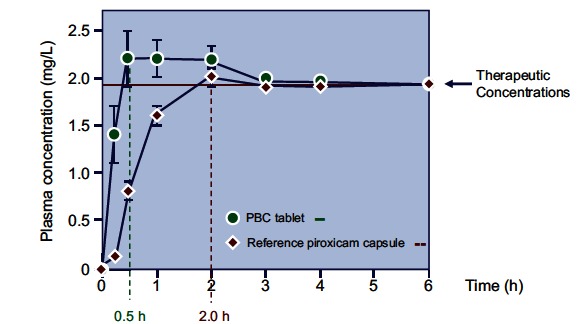
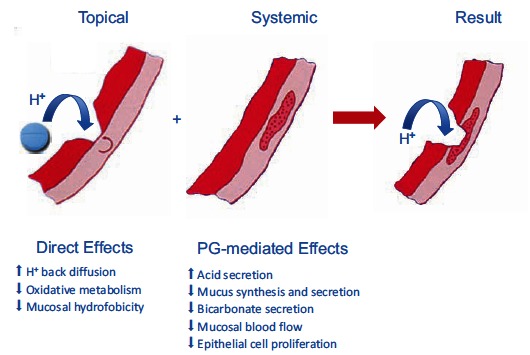
Although NSAIDs are very effective drugs, their use is associated with a broad spectrum of adverse reactions in the liver, kidney, cardiovascular (CV) system, skin and gut. Gastrointestinal (GI) side effects are the most common and constitute a wide clinical spectrum ranging from dyspepsia, heartburn and abdominal discomfort to more serious events such as peptic ulcer with life-threatening complications of bleeding and perforation. The appreciation that CV risk is also increased further complicates the choices of physicians prescribing anti-inflammatory therapy. Despite prevention strategies should be implemented in patients at risk, gastroprotection is often underused and adherence to treatment is generally poor. A more appealing approach would be therefore to develop drugs that are devoid of or have reduced GI toxicity. Gastro- duodenal mucosa possesses many defensive mechanisms and NSAIDs have a deleterious effect on most of them. This results in a mucosa less able to cope with even a reduced acid load. NSAIDs cause gastro-duodenal damage, by two main mechanisms: a physiochemical disruption of the gastric mucosal barrier and systemic inhibition of gastric mucosal protection, through inhibition of cyclooxygenase (COX, PG endoperoxide G/H synthase) activity of the GI mucosa. However, against a background of COX inhibition by anti-inflammatory doses of NSAIDs, their physicochemical properties, in particular their acidity, underlie the topical effect leading to short-term damage. It has been shown that esterification of acidic NSAIDs suppresses their gastrotoxicity without adversely affecting anti-inflammatory activity. Another way to develop NSAIDs with better GI tolerability is to complex these molecules with cyclodextrins (CDs), giving rise to so-called "inclusion complexes" that can have physical, chemical and biological properties very different from either those of the drug or the cyclodextrin. Complexation of NSAIDs with β-cyclodextrin potentially leads to a more rapid onset of action after oral administration and improved GI tolerability because of minimization of the drug gastric effects. One such drug, piroxicam-β-cyclodextrin (PBC), has been used in Europe for 25 years. Preclinical and clinical pharmacology of PBC do show that the β-cyclodextrin inclusion complex of piroxicam is better tolerated from the upper GI tract than free piroxicam, while retaining all the analgesic and anti-inflammatory properties of the parent compound. In addition, the drug is endowed with a quick absorption rate, which translates into a faster onset of analgesic activity, an effect confirmed in several clinical studies. An analysis of the available trials show that PBC has a GI safety profile, which is better than that displayed by uncomplexed piroxicam. Being an inclusion complex of piroxicam, whose CV safety has been pointed out by several observational studies, PBC should be viewed as a CV safe anti-inflmmatory compound and a GI safer alternative to piroxicam. As a consequence, it should be considered as a useful addition to our therapeutic armamentarium.
尽管 NSAIDs 是非常有效的药物,但它们在肝脏、肾脏、心血管(CV)系统、皮肤和肠道中都与广泛的不良反应有关。胃肠道(GI)副作用是最常见的,构成了广泛的临床谱,从消化不良、烧心和腹部不适到更严重的事件,如消化性溃疡,有危及生命的出血和穿孔等并发症。认识到 CV 风险增加进一步使开处方抗炎治疗的医生的选择复杂化。尽管应该在有风险的患者中实施预防策略,但胃肠保护往往未得到充分利用,治疗的依从性通常很差。因此,开发无或减少 GI 毒性的药物将是一种更有吸引力的方法。胃 - 十二指肠黏膜具有许多防御机制,而 NSAIDs 对它们大多数都有有害影响。这导致黏膜更难以应对即使是减少的酸负荷。NSAIDs 通过两种主要机制引起胃 - 十二指肠损伤:胃黏膜屏障的物理化学破坏和通过抑制胃黏膜环氧化酶(COX,PG 内过氧化物 G/H 合酶)活性的全身抑制。然而,在抗炎剂量的 NSAIDs 抑制 COX 的背景下,它们的物理化学性质,特别是它们的酸度,是导致短期损伤的局部作用的基础。已经表明,酸性 NSAIDs 的酯化可抑制其胃肠毒性,而不影响抗炎活性。另一种开发具有更好 GI 耐受性的 NSAIDs 的方法是将这些分子与环糊精(CDs)络合,产生所谓的“包合物”,其可以具有与药物或环糊精的物理、化学和生物学性质非常不同的性质。NSAIDs 与β-环糊精的络合可能导致口服后更快的作用开始,并由于药物胃作用的最小化而改善 GI 耐受性。一种这样的药物,吡罗昔康-β-环糊精(PBC),已在欧洲使用了 25 年。PBC 的临床前和临床药理学确实表明,与游离吡罗昔康相比,吡罗昔康的β-环糊精包合物在胃上部更耐受,同时保留母体化合物的所有镇痛和抗炎特性。此外,该药物具有快速吸收速率,这转化为更快的镇痛作用开始,这在几项临床研究中得到证实。对现有试验的分析表明,PBC 具有比未络合的吡罗昔康更好的 GI 安全性。作为吡罗昔康的包合物,其 CV 安全性已被几项观察性研究指出,PBC 应被视为一种 CV 安全的抗炎化合物和比吡罗昔康更安全的 GI 替代药物。因此,它应该被视为我们治疗武器库中的一个有用的补充。