Genome Center, University of California Davis, Davis, California, USA.
PLoS One. 2013;8(2):e55913. doi: 10.1371/journal.pone.0055913. Epub 2013 Feb 8.
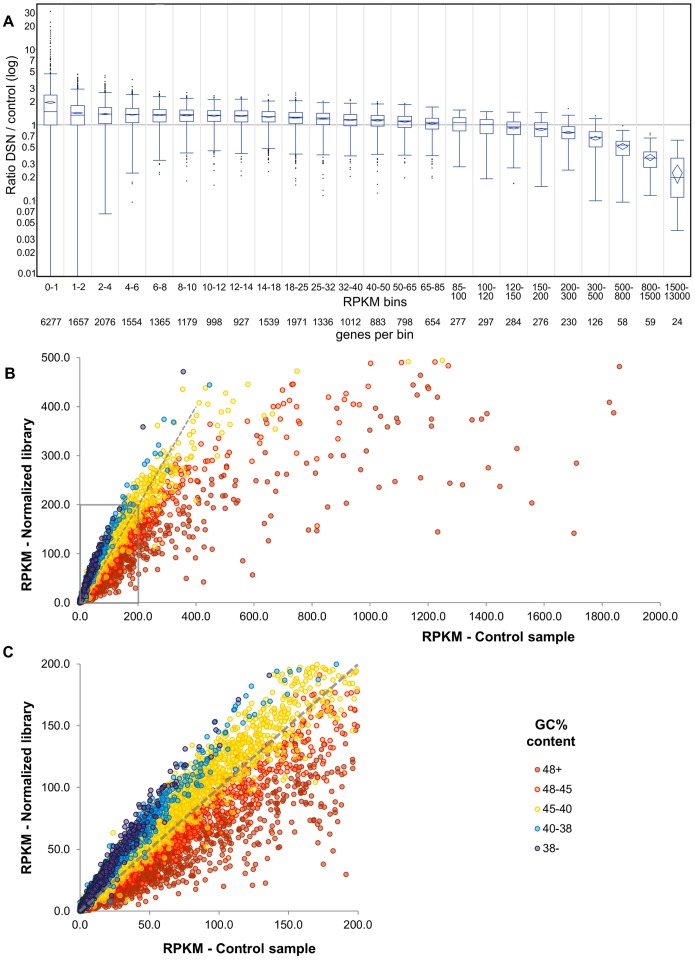
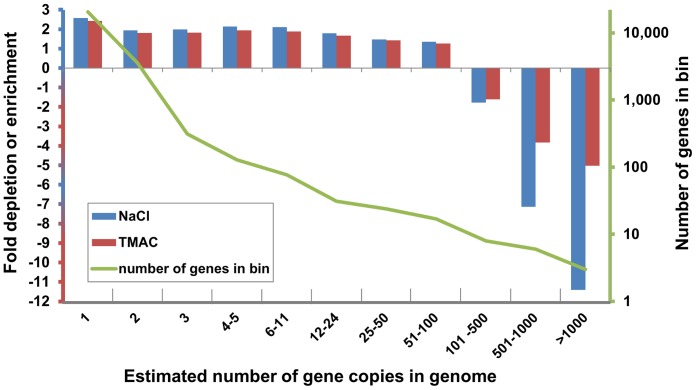
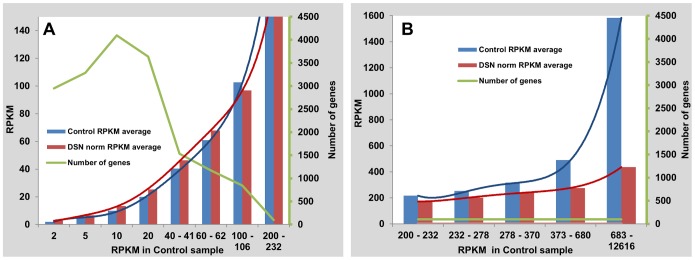
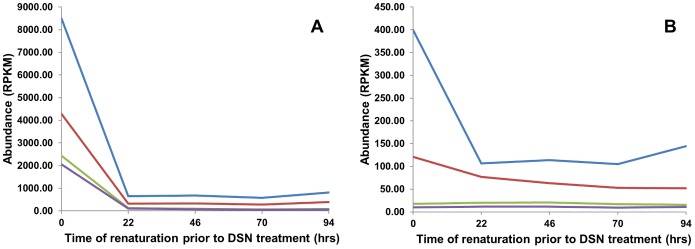
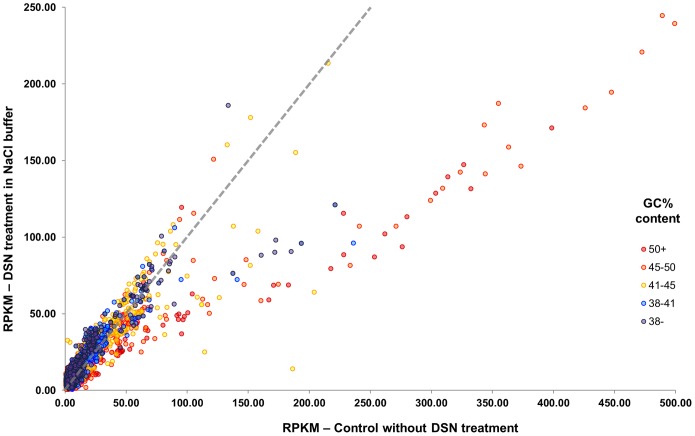
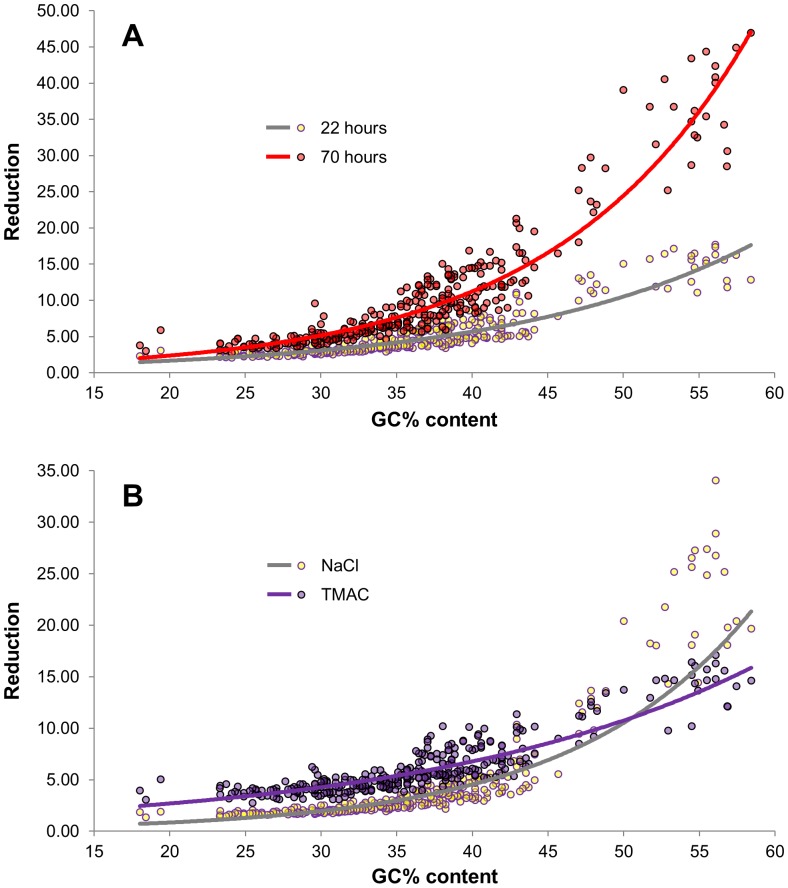
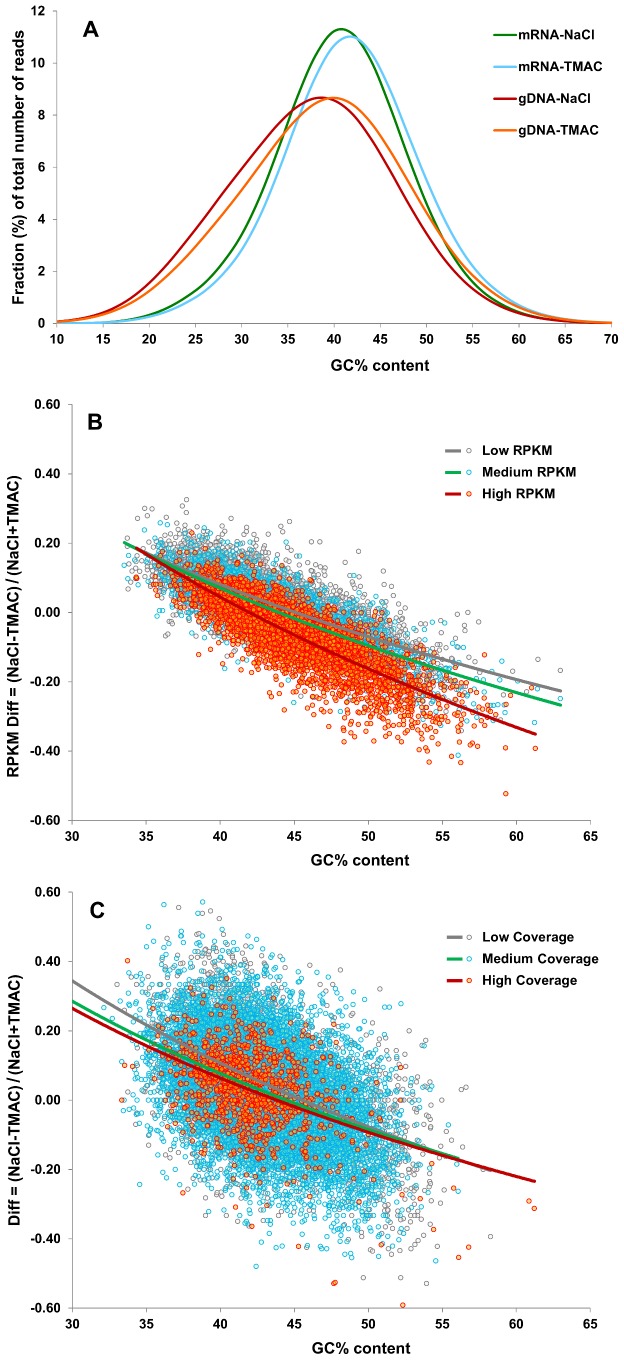
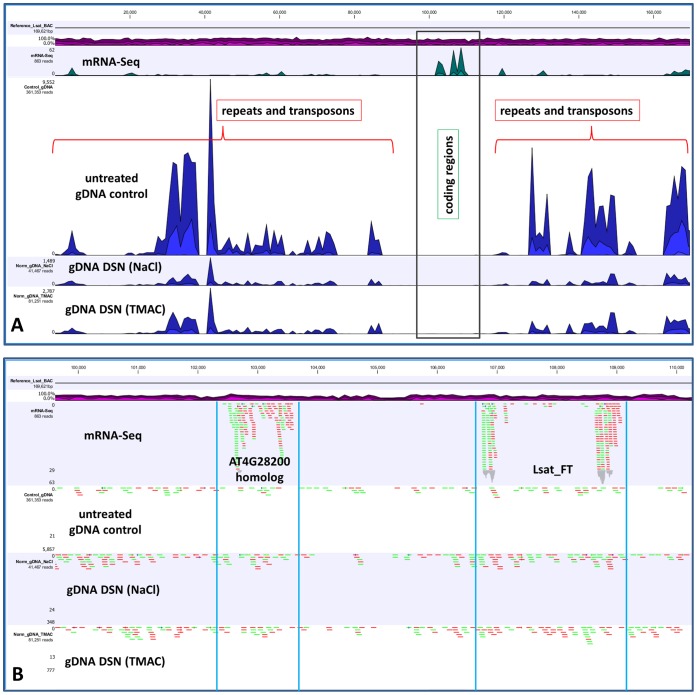
Several applications of high throughput genome and transcriptome sequencing would benefit from a reduction of the high-copy-number sequences in the libraries being sequenced and analyzed, particularly when applied to species with large genomes. We adapted and analyzed the consequences of a method that utilizes a thermostable duplex-specific nuclease for reducing the high-copy components in transcriptomic and genomic libraries prior to sequencing. This reduces the time, cost, and computational effort of obtaining informative transcriptomic and genomic sequence data for both fully sequenced and non-sequenced genomes. It also reduces contamination from organellar DNA in preparations of nuclear DNA. Hybridization in the presence of 3 M tetramethylammonium chloride (TMAC), which equalizes the rates of hybridization of GC and AT nucleotide pairs, reduced the bias against sequences with high GC content. Consequences of this method on the reduction of high-copy and enrichment of low-copy sequences are reported for Arabidopsis and lettuce.
高通量基因组和转录组测序的几种应用将受益于减少正在测序和分析的文库中的高拷贝数序列,尤其是在应用于基因组较大的物种时。我们对一种方法进行了改编和分析,该方法利用热稳定的双链特异性核酸酶在测序前减少转录组和基因组文库中的高拷贝成分。这减少了获得全序列和非序列基因组的信息丰富的转录组和基因组序列数据的时间、成本和计算工作量。它还减少了核 DNA 制备中来自细胞器 DNA 的污染。在存在 3 M 四甲基氯化铵 (TMAC) 的情况下进行杂交,使 GC 和 AT 核苷酸对的杂交速率相等,减少了对高 GC 含量序列的偏见。本文报告了该方法对拟南芥和生菜中高拷贝序列减少和低拷贝序列富集的影响。