Department of Forest Ecosystems and Society, Oregon State University, Corvallis, Oregon 97331, USA.
BMC Genomics. 2013 Feb 28;14:137. doi: 10.1186/1471-2164-14-137.
Douglas-fir (Pseudotsuga menziesii), one of the most economically and ecologically important tree species in the world, also has one of the largest tree breeding programs. Although the coastal and interior varieties of Douglas-fir (vars. menziesii and glauca) are native to North America, the coastal variety is also widely planted for timber production in Europe, New Zealand, Australia, and Chile. Our main goal was to develop a SNP resource large enough to facilitate genomic selection in Douglas-fir breeding programs. To accomplish this, we developed a 454-based reference transcriptome for coastal Douglas-fir, annotated and evaluated the quality of the reference, identified putative SNPs, and then validated a sample of those SNPs using the Illumina Infinium genotyping platform.
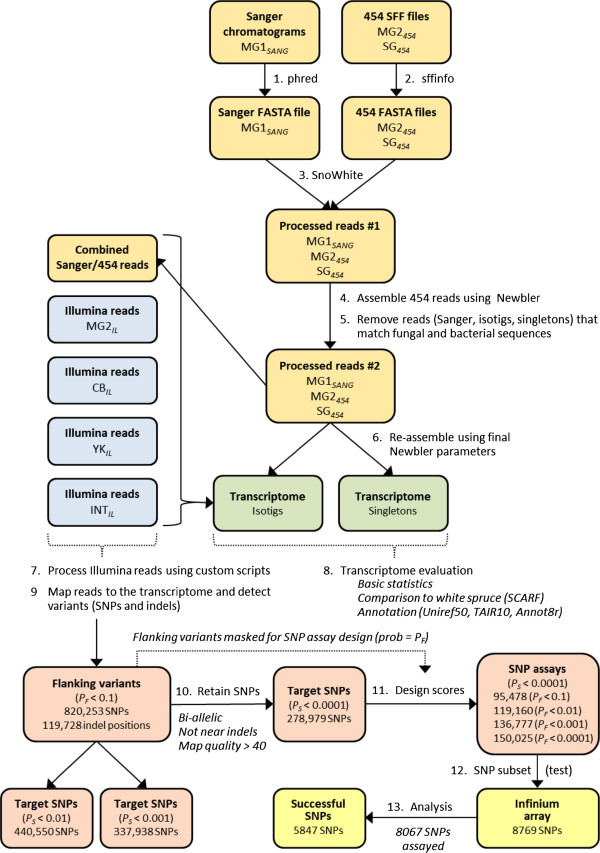
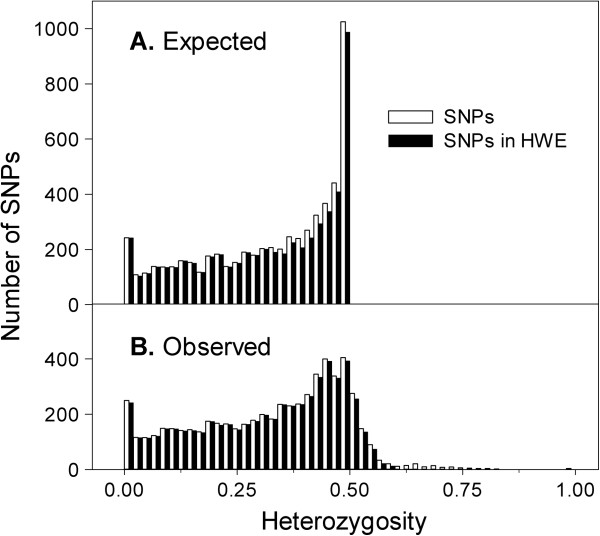
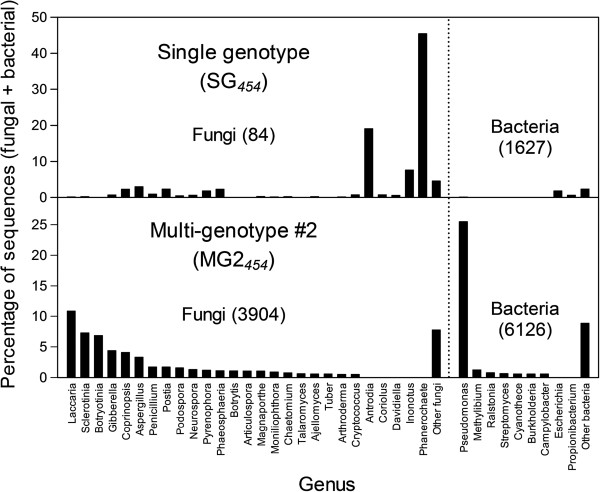
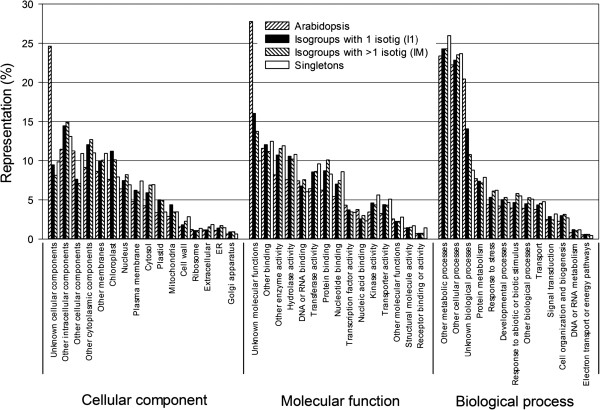
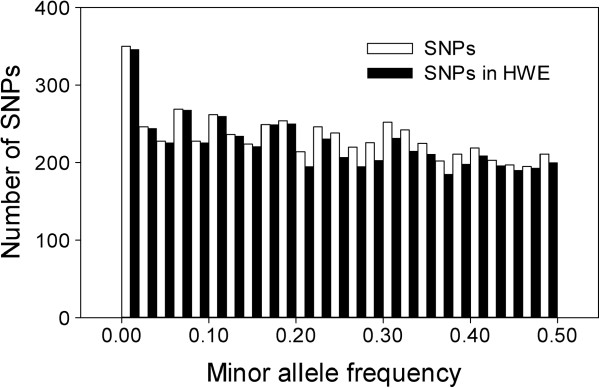
We assembled a reference transcriptome consisting of 25,002 isogroups (unique gene models) and 102,623 singletons from 2.76 million 454 and Sanger cDNA sequences from coastal Douglas-fir. We identified 278,979 unique SNPs by mapping the 454 and Sanger sequences to the reference, and by mapping four datasets of Illumina cDNA sequences from multiple seed sources, genotypes, and tissues. The Illumina datasets represented coastal Douglas-fir (64.00 and 13.41 million reads), interior Douglas-fir (80.45 million reads), and a Yakima population similar to interior Douglas-fir (8.99 million reads). We assayed 8067 SNPs on 260 trees using an Illumina Infinium SNP genotyping array. Of these SNPs, 5847 (72.5%) were called successfully and were polymorphic.
Based on our validation efficiency, our SNP database may contain as many as ~200,000 true SNPs, and as many as ~69,000 SNPs that could be genotyped at ~20,000 gene loci using an Infinium II array-more SNPs than are needed to use genomic selection in tree breeding programs. Ultimately, these genomic resources will enhance Douglas-fir breeding and allow us to better understand landscape-scale patterns of genetic variation and potential responses to climate change.
花旗松(Pseudotsuga menziesii)是世界上经济和生态上最重要的树种之一,也是树木育种计划规模最大的树种之一。尽管沿海和内陆花旗松(var. menziesii 和 glauca)原产于北美,但沿海品种也广泛种植于欧洲、新西兰、澳大利亚和智利,用于木材生产。我们的主要目标是开发一个足够大的 SNP 资源,以促进花旗松育种计划中的基因组选择。为了实现这一目标,我们为沿海花旗松开发了一个基于 454 的参考转录组,对参考转录组进行注释和质量评估,鉴定出可能的 SNP,然后使用 Illumina Infinium 基因分型平台对这些 SNP 的样本进行验证。
我们组装了一个参考转录组,该转录组由来自沿海花旗松的 276 万 454 和 Sanger cDNA 序列的 25002 个同基因簇(独特的基因模型)和 102623 个单核苷酸组成。我们通过将 454 和 Sanger 序列映射到参考序列上,以及通过映射来自多个种子来源、基因型和组织的四个 Illumina cDNA 数据集,鉴定出 278979 个独特的 SNP。Illumina 数据集代表沿海花旗松(6400 万和 1341 万条reads)、内陆花旗松(8045 万条reads)和类似于内陆花旗松的 Yakima 种群(899 万条reads)。我们使用 Illumina Infinium SNP 基因分型阵列在 260 棵树上检测了 8067 个 SNP。在这些 SNP 中,5847 个(72.5%)成功地被检测到并具有多态性。
根据我们的验证效率,我们的 SNP 数据库可能包含多达 200000 个真实 SNP,以及多达 69000 个 SNP,可以在 20000 个基因座上使用 Infinium II 阵列进行基因分型——这比树木育种计划中使用基因组选择所需的 SNP 多。最终,这些基因组资源将增强花旗松的育种,并使我们能够更好地理解景观尺度的遗传变异模式和对气候变化的潜在反应。