College of Agriculture and Biotechnology, China Agricultural University, Beijing, China.
PLoS One. 2013;8(2):e57686. doi: 10.1371/journal.pone.0057686. Epub 2013 Feb 28.
Celery is an increasing popular vegetable species, but limited transcriptome and genomic data hinder the research to it. In addition, a lack of celery molecular markers limits the process of molecular genetic breeding. High-throughput transcriptome sequencing is an efficient method to generate a large transcriptome sequence dataset for gene discovery, molecular marker development and marker-assisted selection breeding.
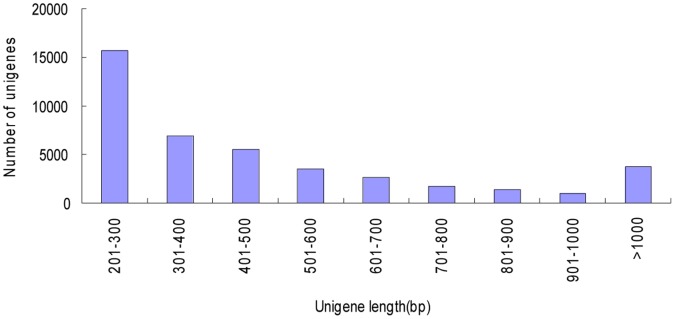
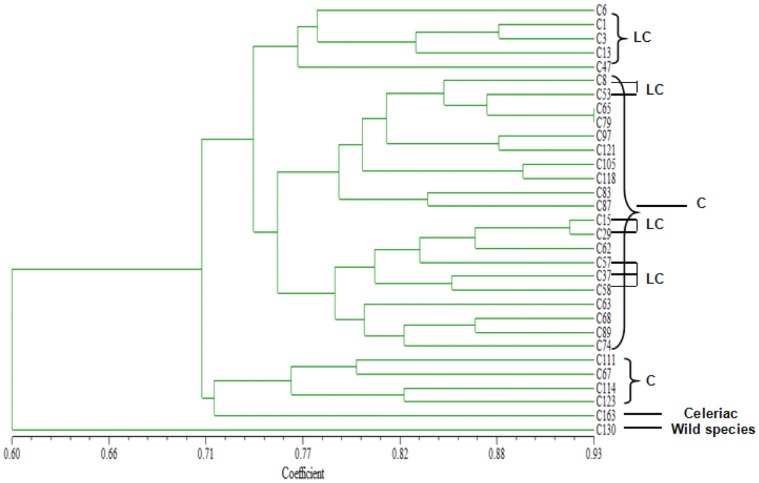
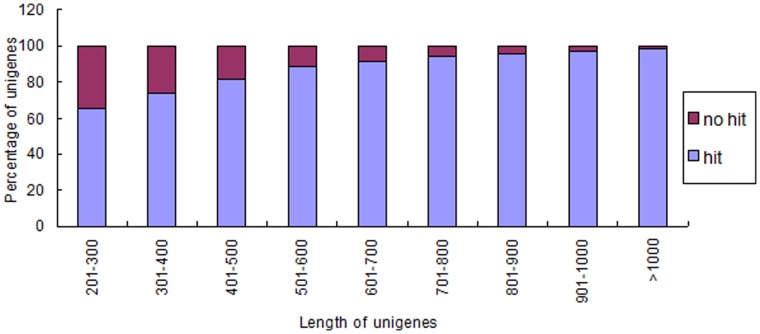
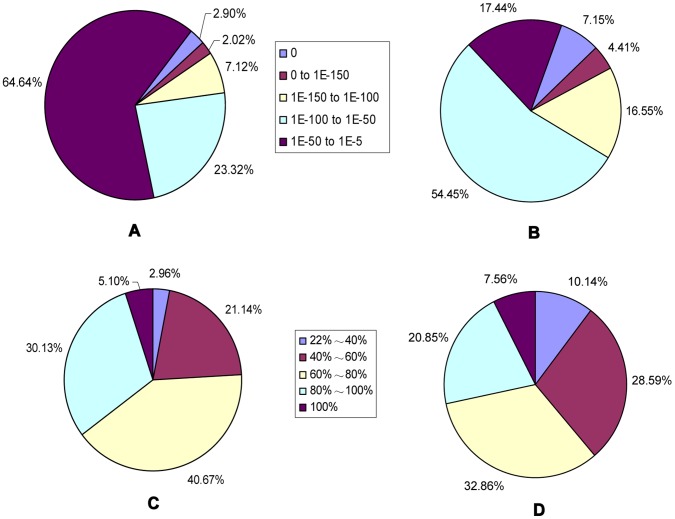
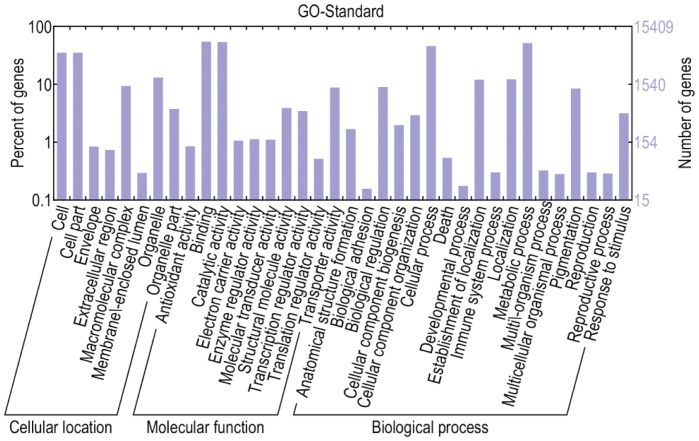
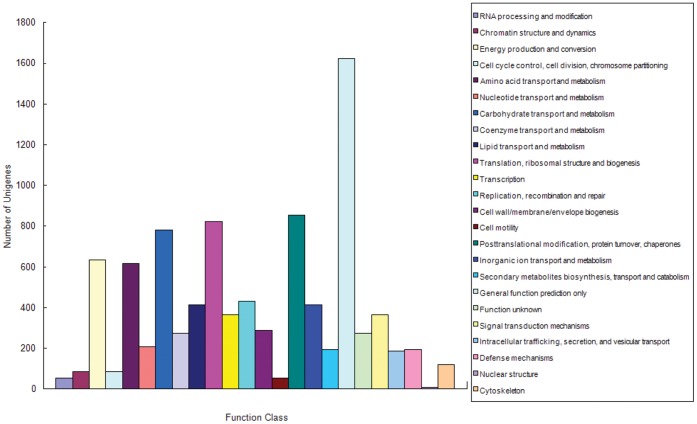
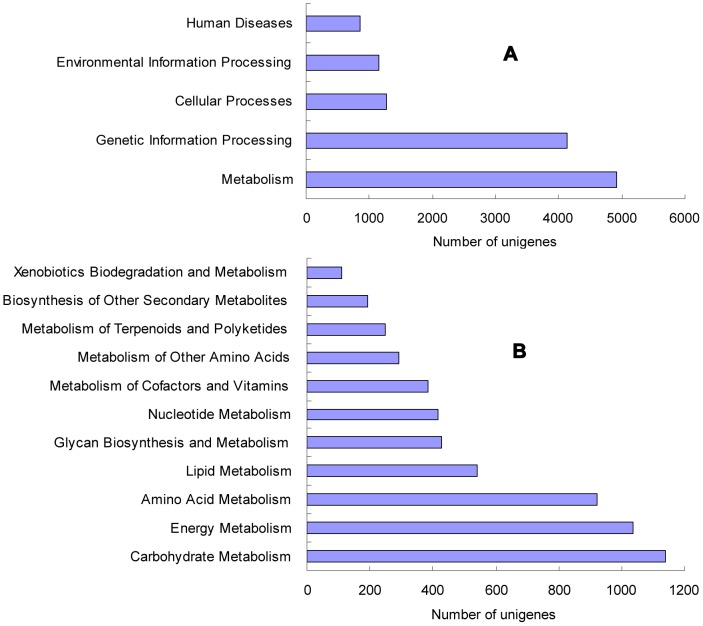
Celery transcriptomes from four tissues were sequenced using Illumina paired-end sequencing technology. De novo assembling was performed to generate a collection of 42,280 unigenes (average length of 502.6 bp) that represent the first transcriptome of the species. 78.43% and 48.93% of the unigenes had significant similarity with proteins in the National Center for Biotechnology Information (NCBI) non-redundant protein database (Nr) and Swiss-Prot database respectively, and 10,473 (24.77%) unigenes were assigned to Clusters of Orthologous Groups (COG). 21,126 (49.97%) unigenes harboring Interpro domains were annotated, in which 15,409 (36.45%) were assigned to Gene Ontology(GO) categories. Additionally, 7,478 unigenes were mapped onto 228 pathways using the Kyoto Encyclopedia of Genes and Genomes Pathway database (KEGG). Large numbers of simple sequence repeats (SSRs) were indentified, and then the rate of successful amplication and polymorphism were investigated among 31 celery accessions.
This study demonstrates the feasibility of generating a large scale of sequence information by Illumina paired-end sequencing and efficient assembling. Our results provide a valuable resource for celery research. The developed molecular markers are the foundation of further genetic linkage analysis and gene localization, and they will be essential to accelerate the process of breeding.
芹菜是一种日益流行的蔬菜物种,但有限的转录组和基因组数据阻碍了对其的研究。此外,缺乏芹菜分子标记限制了分子遗传育种的进程。高通量转录组测序是一种高效的方法,可以生成大量的转录组序列数据集,用于基因发现、分子标记开发和标记辅助选择育种。
使用 Illumina 配对末端测序技术对来自四个组织的芹菜转录组进行测序。通过从头组装生成了一个包含 42280 个基因(平均长度为 502.6bp)的集合,代表该物种的第一个转录组。78.43%和 48.93%的基因与美国国家生物技术信息中心(NCBI)非冗余蛋白质数据库(Nr)和瑞士 - 普洛特数据库中的蛋白质具有显著相似性,10473 个(24.77%)基因被分配到同源基因簇(COG)。21126 个(49.97%)含有 Interpro 结构域的基因被注释,其中 15409 个(36.45%)被分配到基因本体论(GO)类别。此外,使用京都基因与基因组百科全书途径数据库(KEGG)将 7478 个基因映射到 228 个途径上。鉴定了大量的简单序列重复(SSR),然后调查了 31 个芹菜品种的成功扩增和多态性率。
本研究证明了通过 Illumina 配对末端测序和高效组装生成大规模序列信息的可行性。我们的结果为芹菜研究提供了有价值的资源。开发的分子标记是进一步遗传连锁分析和基因定位的基础,它们对于加速育种进程至关重要。