Department of Biology, San Diego State University, San Diego, California, USA.
PLoS One. 2013;8(3):e58404. doi: 10.1371/journal.pone.0058404. Epub 2013 Mar 15.
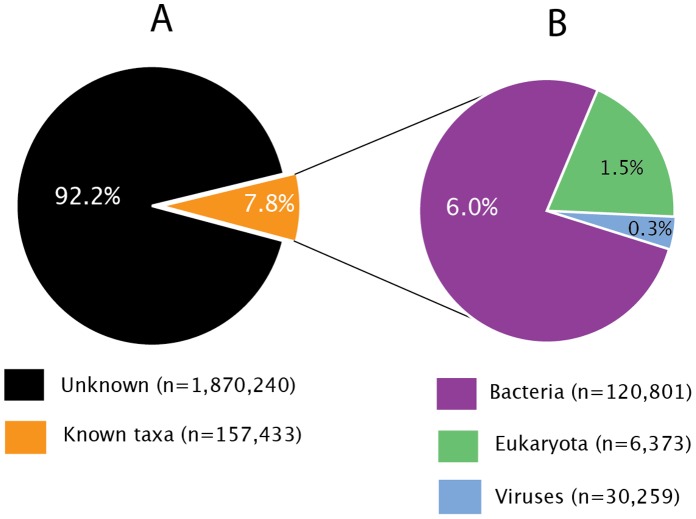
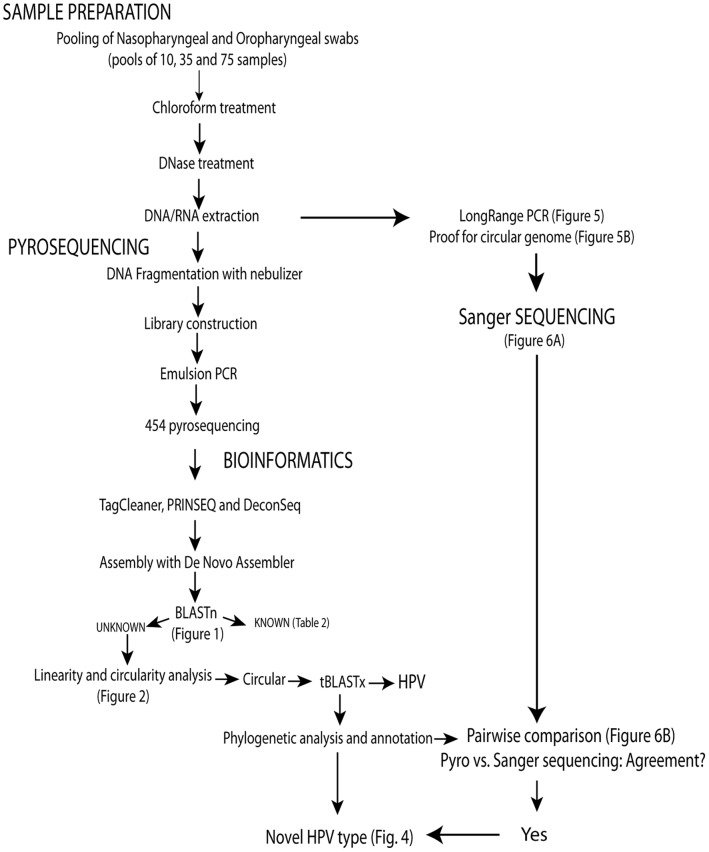
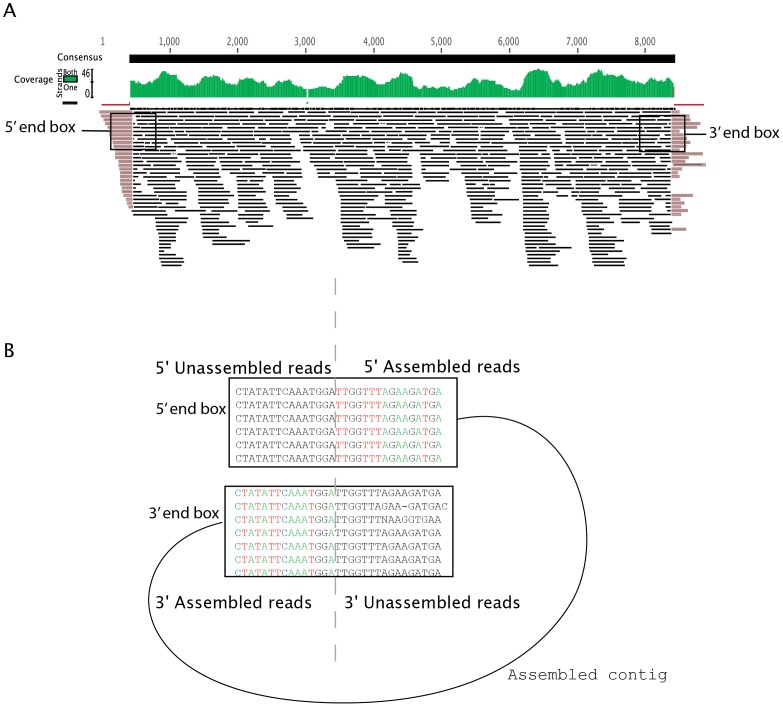
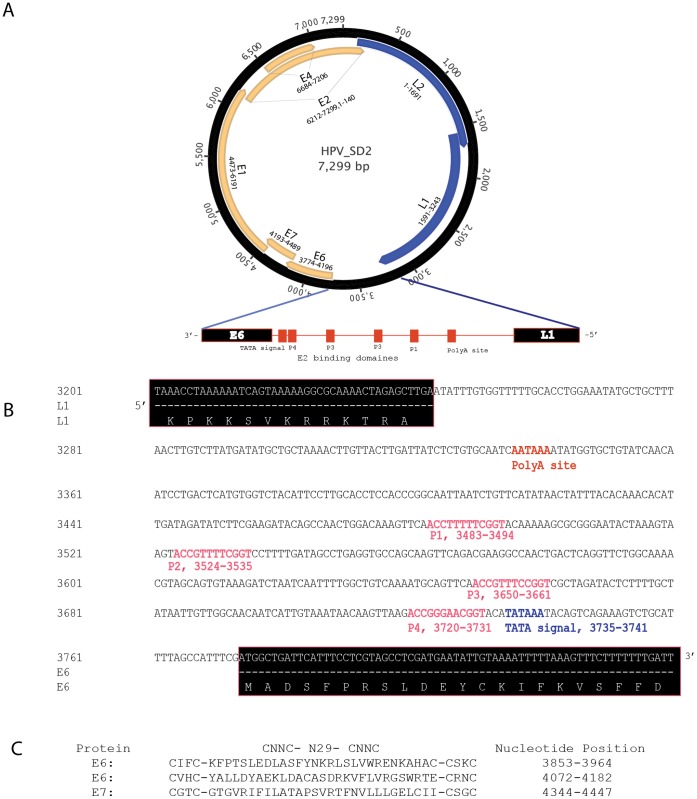
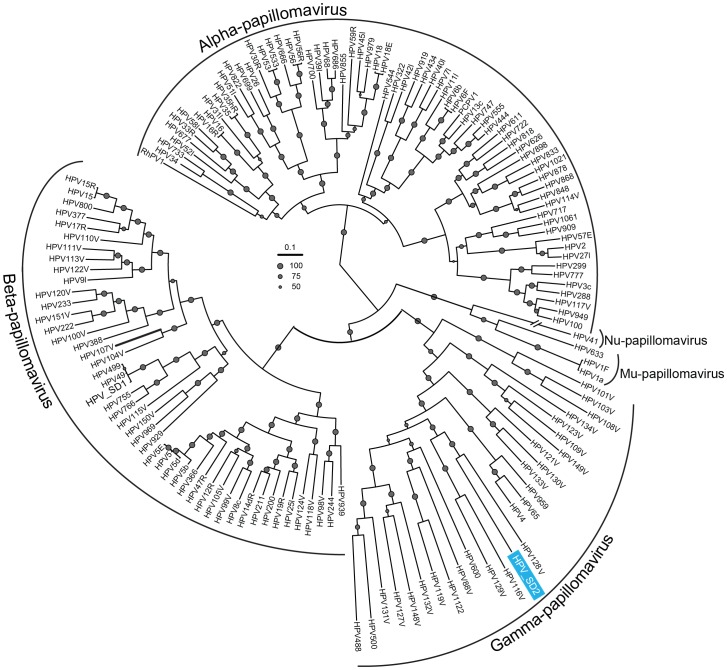
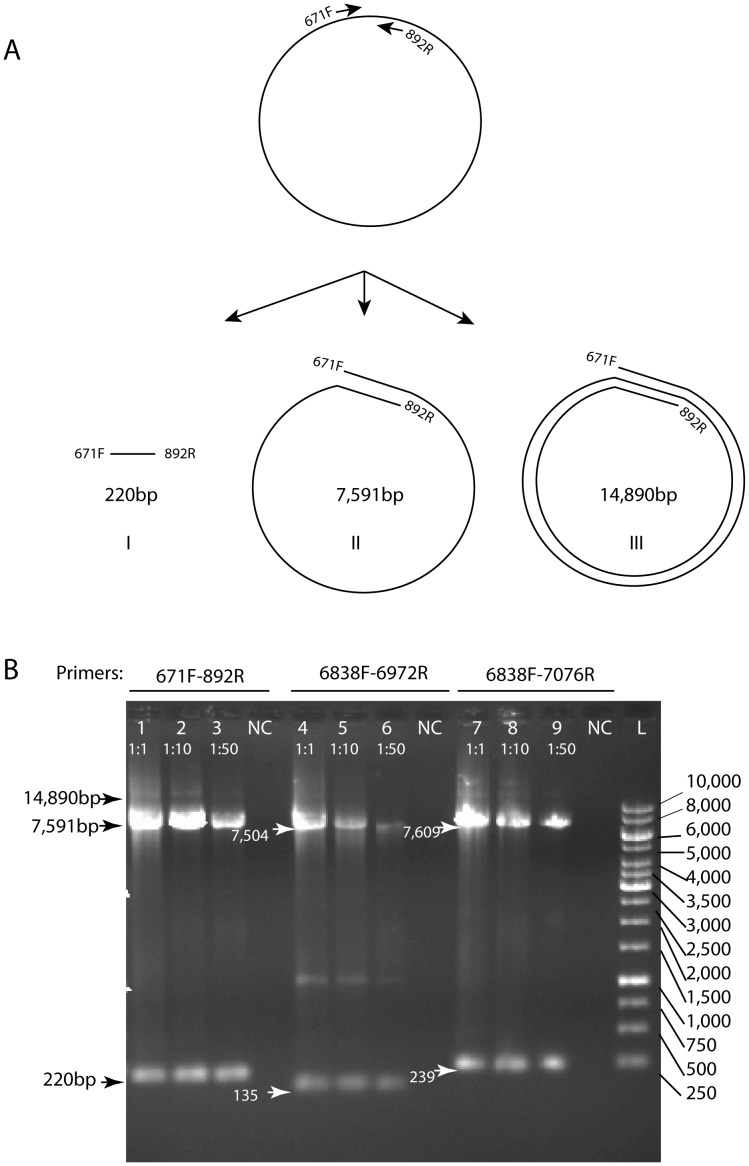
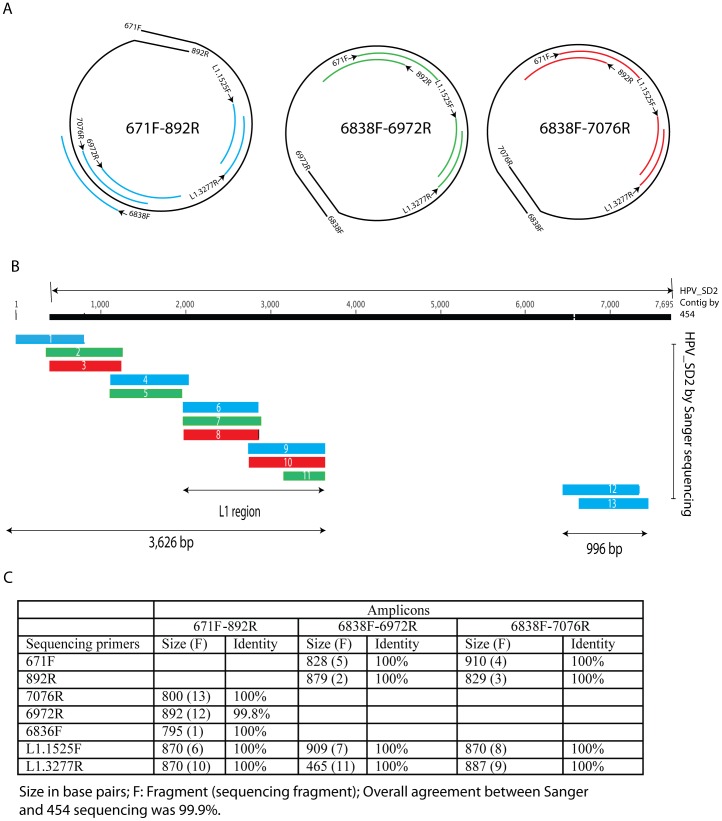
As part of a virus discovery investigation using a metagenomic approach, a highly divergent novel Human papillomavirus type was identified in pooled convenience nasal/oropharyngeal swab samples collected from patients with febrile respiratory illness. Phylogenetic analysis of the whole genome and the L1 gene reveals that the new HPV identified in this study clusters with previously described gamma papillomaviruses, sharing only 61.1% (whole genome) and 63.1% (L1) sequence identity with its closest relative in the Papillomavirus episteme (PAVE) database. This new virus was named HPV_SD2 pending official classification. The complete genome of HPV-SD2 is 7,299 bp long (36.3% G/C) and contains 7 open reading frames (L2, L1, E6, E7, E1, E2 and E4) and a non-coding long control region (LCR) between L1 and E6. The metagenomic procedures, coupled with the bioinformatic methods described herein are well suited to detect small circular genomes such as those of human papillomaviruses.
在一项使用宏基因组方法进行病毒发现调查的过程中,从患有发热性呼吸道疾病的患者采集的混合便利鼻/咽拭子样本中鉴定出一种高度分化的新型人乳头瘤病毒。全基因组和 L1 基因的系统发育分析表明,本研究中鉴定出的新型 HPV 与先前描述的γ乳头瘤病毒聚为一簇,与 Papillomavirus episteme (PAVE) 数据库中最接近的亲缘关系仅有 61.1%(全基因组)和 63.1%(L1)的序列同一性。该新病毒被命名为 HPV_SD2,等待正式分类。HPV-SD2 的完整基因组长 7299bp(36.3%G/C),包含 7 个开放阅读框(L2、L1、E6、E7、E1、E2 和 E4)和一个位于 L1 和 E6 之间的非编码长控制区(LCR)。本文所述的宏基因组程序和生物信息学方法非常适合检测小的环状基因组,如人乳头瘤病毒。