Department of Physiology and Medical Physics, Royal College of Surgeons in Ireland, Dublin, Ireland.
Cell Death Dis. 2013 Apr 25;4(4):e606. doi: 10.1038/cddis.2013.136.
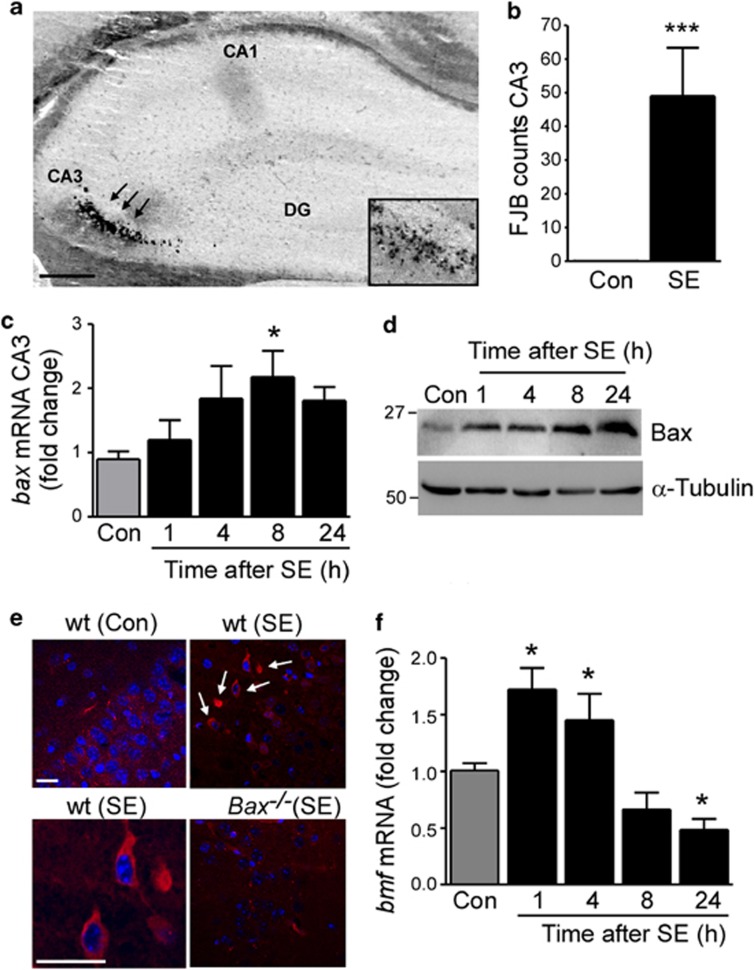
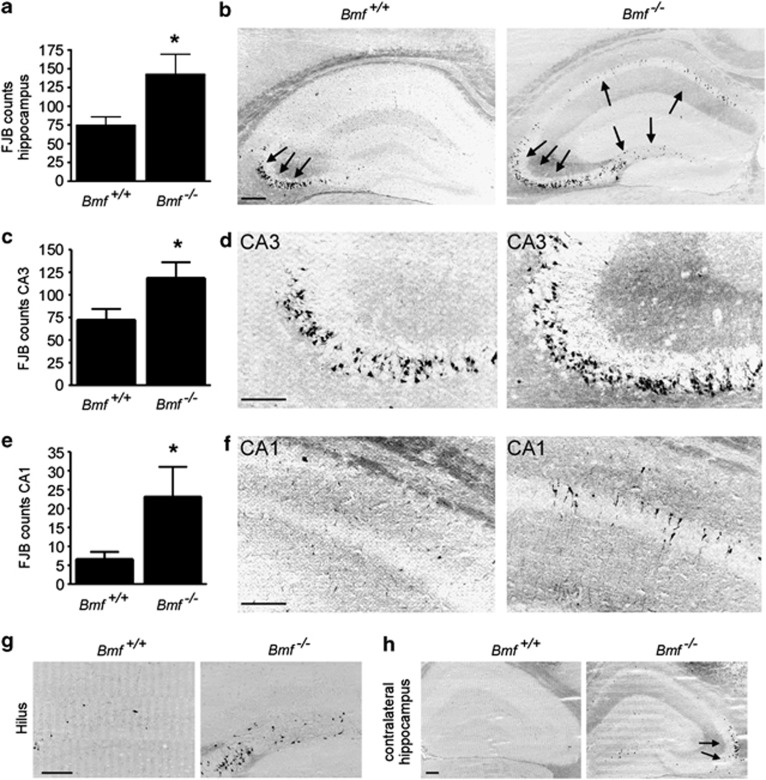
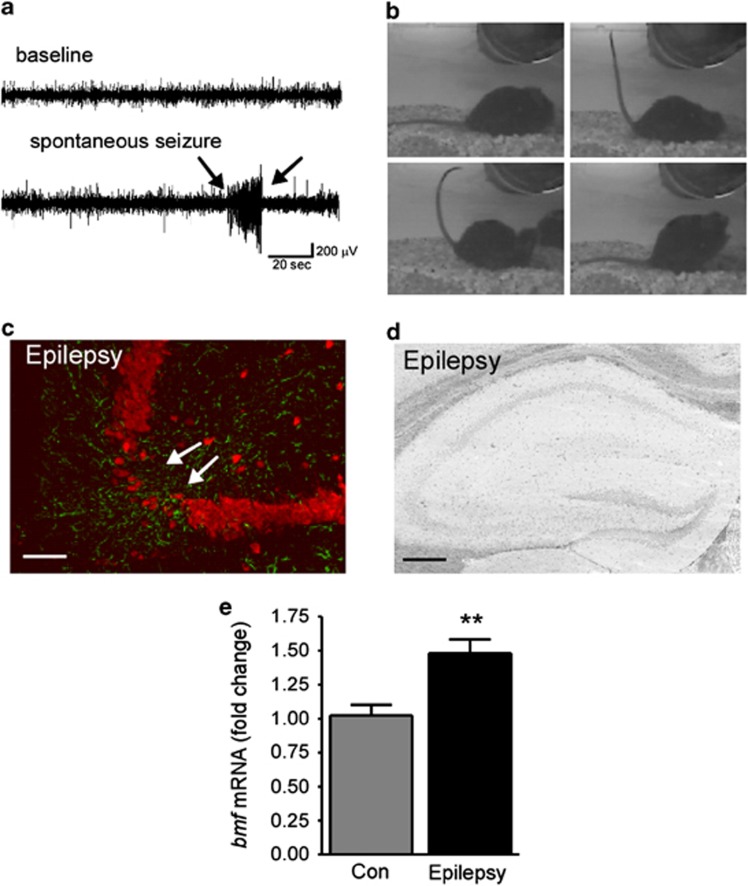
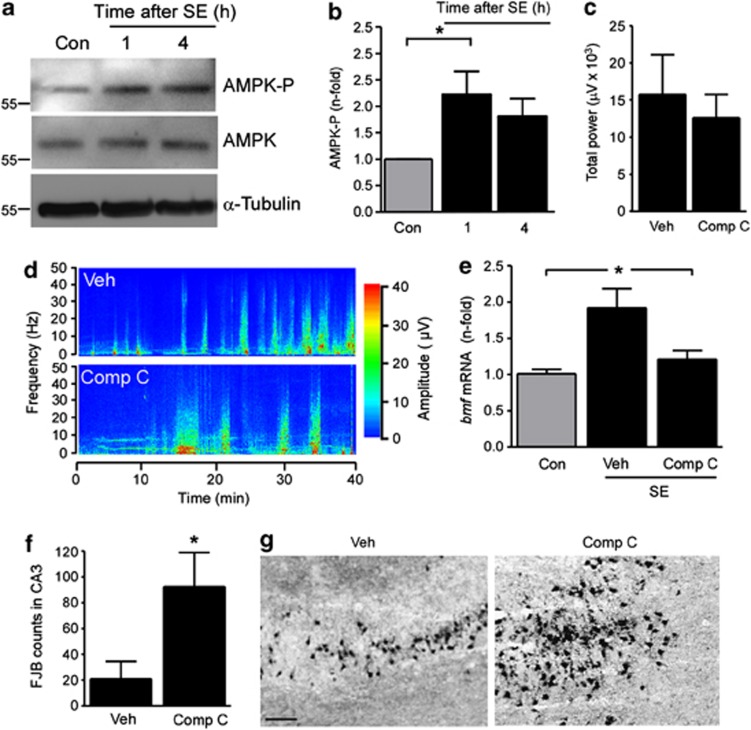
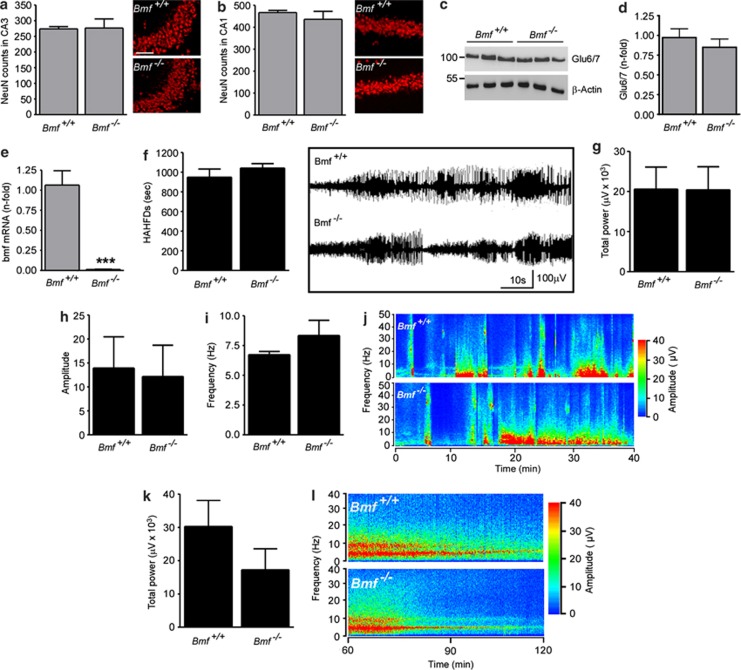
Prolonged seizures (status epilepticus, SE) can cause neuronal death within brain regions such as the hippocampus. This may contribute to impairments in cognitive functioning and trigger or exacerbate epilepsy. Seizure-induced neuronal death is mediated, at least in part, by apoptosis-associated signaling pathways. Indeed, mice lacking certain members of the potently proapoptotic BH3-only subfamily of Bcl-2 proteins are protected against hippocampal damage caused by status epilepticus. The recently identified BH3-only protein Bcl-2-modifying factor (Bmf) normally interacts with the cytoskeleton, but upon certain cellular stresses, such as loss of extracellular matrix adhesion or energy crisis, Bmf relocalizes to mitochondria, where it can promote Bax activation and mitochondrial dysfunction. Although Bmf has been widely reported in the hematopoietic system to exert a proapoptotic effect, no studies have been undertaken in models of neurological disorders. To examine whether Bmf is important for seizure-induced neuronal death, we studied Bmf induction after prolonged seizures induced by intra-amygdala kainic acid (KA) in mice, and examined the effect of Bmf-deficiency on seizures and damage caused by SE. Seizures triggered an early (1-8 h) transcriptional activation and accumulation of Bax in the cell death-susceptible hippocampal CA3 subfield. Bmf mRNA was biphasically upregulated beginning at 1 h after SE and returning to normal by 8 h, while again being found elevated in the hippocampus of epileptic mice. Bmf upregulation was prevented by Compound C, an inhibitor of adenosine monophosphate-activated protein kinase, indicating Bmf expression may be induced in response to bioenergetic stress. Bmf-deficient mice showed normal sensitivity to the convulsant effects of KA, but, surprisingly, displayed significantly more neuronal death in the hippocampal CA1 and CA3 subfields after SE. These are the first studies investigating Bmf in a model of neurologic injury, and suggest that Bmf may protect neurons against seizure-induced neuronal death in vivo.
长时间的癫痫发作(癫痫持续状态,SE)可导致海马等脑区神经元死亡。这可能导致认知功能障碍,并引发或加重癫痫。癫痫发作诱导的神经元死亡至少部分是由凋亡相关信号通路介导的。事实上,缺乏某些促凋亡 BH3 仅亚家族 Bcl-2 蛋白成员的小鼠可免受癫痫持续状态引起的海马损伤。最近发现的 BH3 仅蛋白 Bcl-2 修饰因子(Bmf)通常与细胞骨架相互作用,但在某些细胞应激下,如细胞外基质粘附丧失或能量危机,Bmf 重新定位到线粒体,在那里它可以促进 Bax 激活和线粒体功能障碍。尽管 Bmf 在造血系统中被广泛报道具有促凋亡作用,但尚未在神经疾病模型中进行研究。为了研究 Bmf 是否对癫痫发作诱导的神经元死亡很重要,我们研究了在小鼠中通过内侧杏仁核海人酸(KA)诱导长时间癫痫发作后的 Bmf 诱导,并研究了 Bmf 缺失对 SE 引起的癫痫发作和损伤的影响。癫痫发作引发了细胞死亡易感的海马 CA3 亚区中 Bax 的早期(1-8 小时)转录激活和积累。Bmf mRNA 呈双相上调,在 SE 后 1 小时开始,并在 8 小时时恢复正常,而在癫痫小鼠的海马中再次升高。用 AMPK 抑制剂 Compound C 可阻止 Bmf 上调,表明 Bmf 表达可能是对生物能量应激的反应。Bmf 缺失小鼠对 KA 的致惊厥作用表现出正常的敏感性,但令人惊讶的是,在 SE 后,海马 CA1 和 CA3 亚区的神经元死亡明显增加。这些是首次在神经损伤模型中研究 Bmf 的研究,表明 Bmf 可能在体内保护神经元免受癫痫发作诱导的神经元死亡。