Department of Physiology, Development and Neuroscience, University of Cambridge, Cambridge, United Kingdom.
PLoS One. 2013 Apr 16;8(4):e60847. doi: 10.1371/journal.pone.0060847. Print 2013.
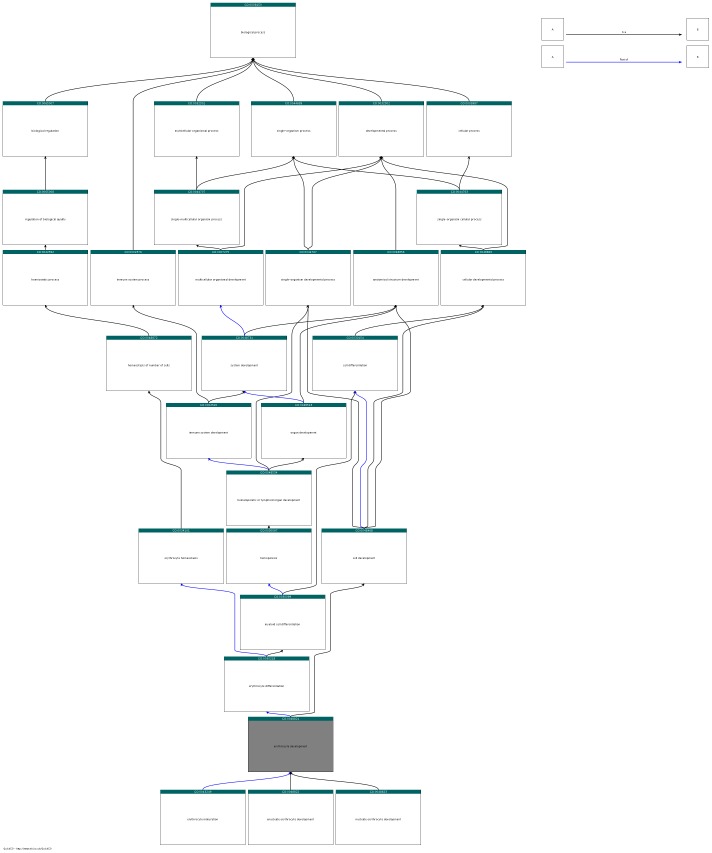
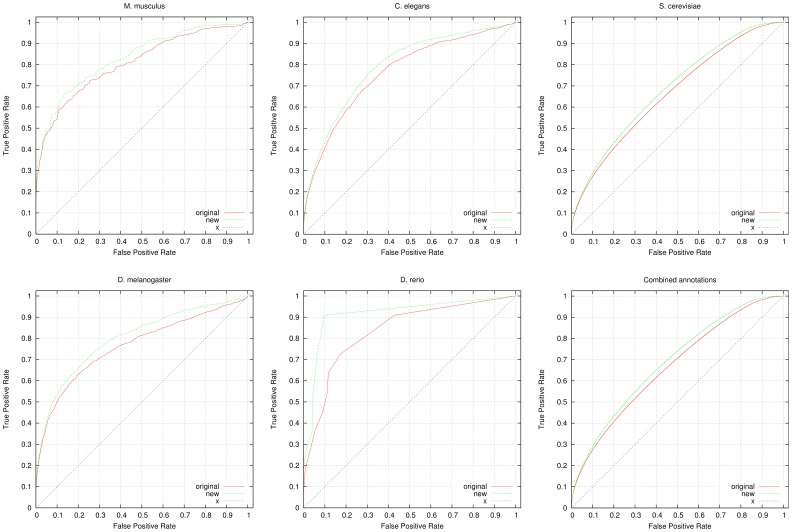
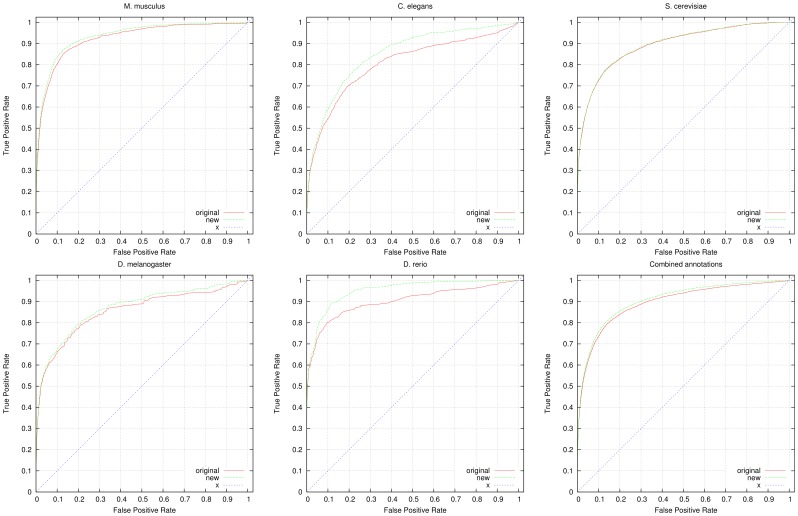
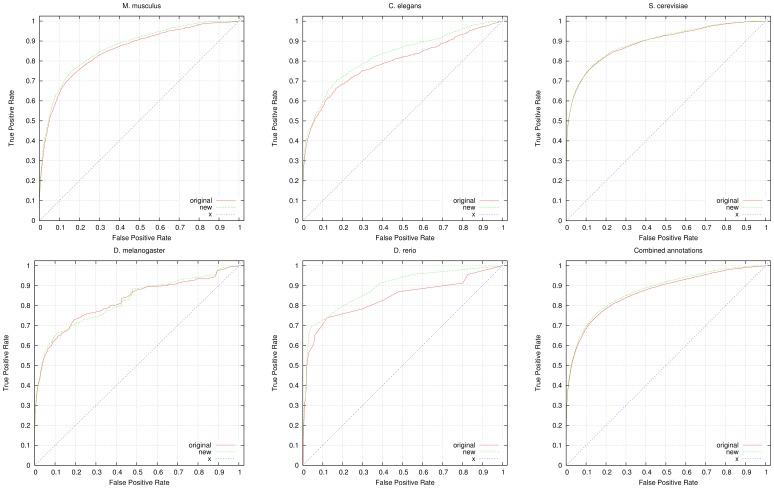
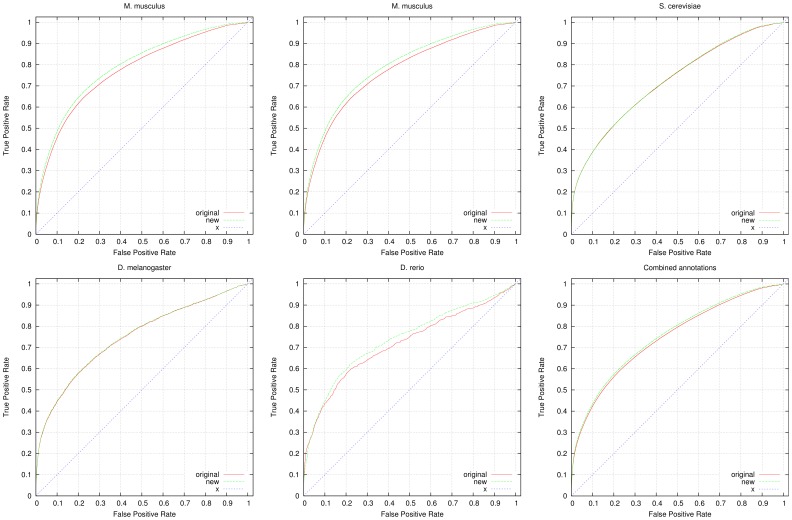
High-throughput phenotyping projects in model organisms have the potential to improve our understanding of gene functions and their role in living organisms. We have developed a computational, knowledge-based approach to automatically infer gene functions from phenotypic manifestations and applied this approach to yeast (Saccharomyces cerevisiae), nematode worm (Caenorhabditis elegans), zebrafish (Danio rerio), fruitfly (Drosophila melanogaster) and mouse (Mus musculus) phenotypes. Our approach is based on the assumption that, if a mutation in a gene [Formula: see text] leads to a phenotypic abnormality in a process [Formula: see text], then [Formula: see text] must have been involved in [Formula: see text], either directly or indirectly. We systematically analyze recorded phenotypes in animal models using the formal definitions created for phenotype ontologies. We evaluate the validity of the inferred functions manually and by demonstrating a significant improvement in predicting genetic interactions and protein-protein interactions based on functional similarity. Our knowledge-based approach is generally applicable to phenotypes recorded in model organism databases, including phenotypes from large-scale, high throughput community projects whose primary mode of dissemination is direct publication on-line rather than in the literature.
在模式生物中进行高通量表型分析项目有可能增进我们对基因功能及其在生物体中作用的理解。我们开发了一种基于计算和知识的方法,能够自动从表型表现推断基因功能,并将该方法应用于酵母(酿酒酵母)、线虫(秀丽隐杆线虫)、斑马鱼(Danio rerio)、果蝇(Drosophila melanogaster)和小鼠(Mus musculus)的表型中。我们的方法基于这样一种假设,即如果一个基因 [Formula: see text] 的突变导致一个过程 [Formula: see text] 中的表型异常,那么 [Formula: see text] 必然直接或间接地参与了该过程。我们使用为表型本体论创建的形式定义,系统地分析动物模型中的记录表型。我们通过手动评估和证明基于功能相似性预测遗传相互作用和蛋白质-蛋白质相互作用的能力显著提高,来验证推断功能的有效性。我们的基于知识的方法通常适用于模式生物数据库中记录的表型,包括来自大规模、高通量社区项目的表型,这些项目的主要传播方式是直接在线发布,而不是在文献中发布。