Molecular and Computational Biology Program, University of Southern California, Los Angeles, CA 90089, USA.
J Cell Biol. 2013 Apr 29;201(3):373-83. doi: 10.1083/jcb.201208060.
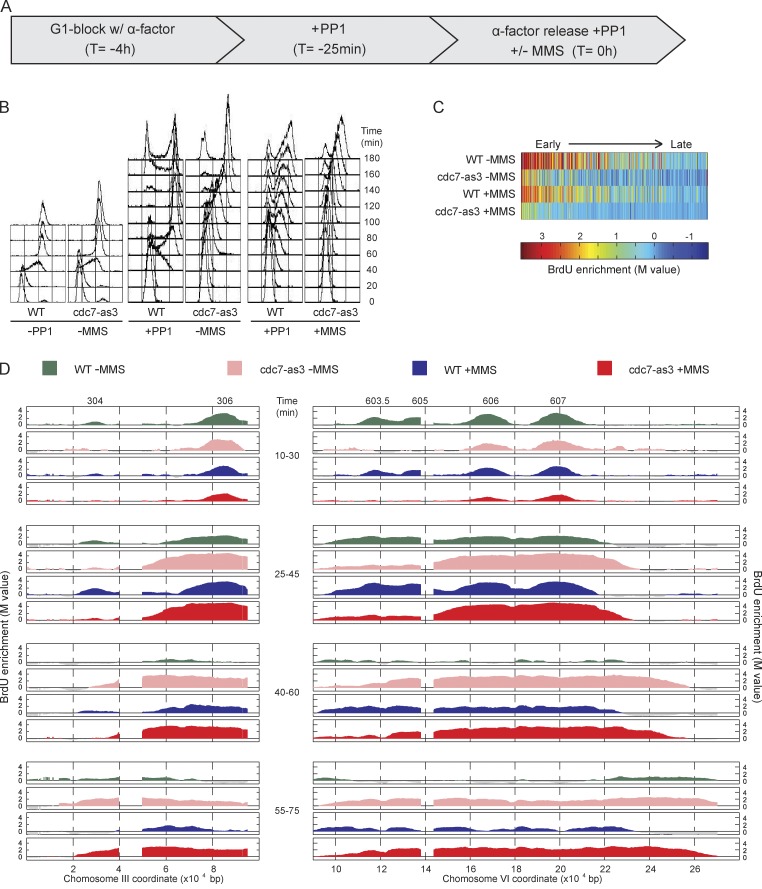
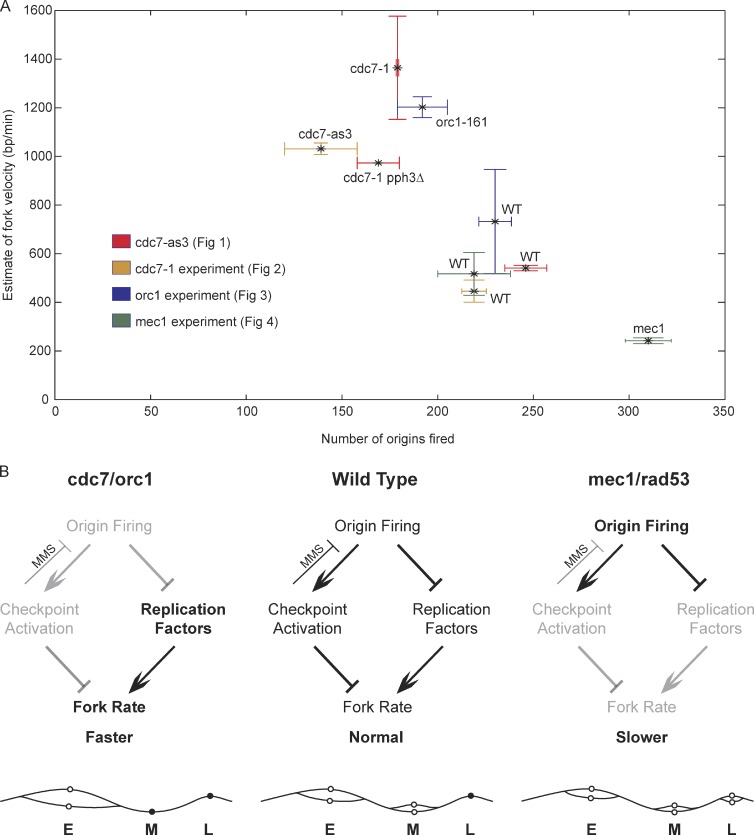
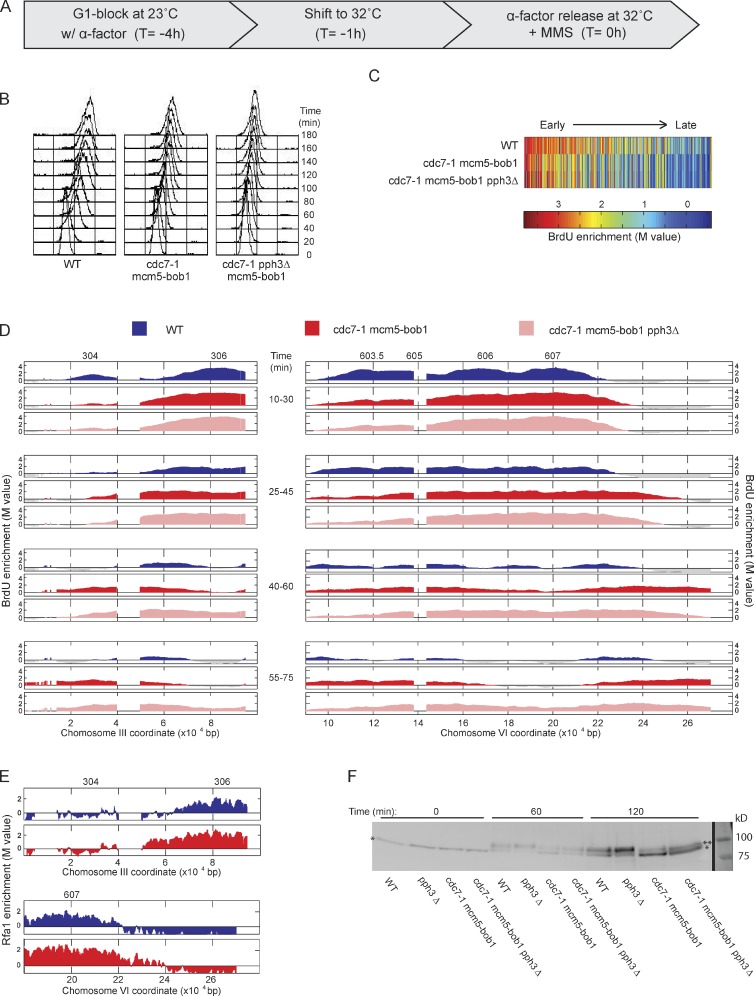
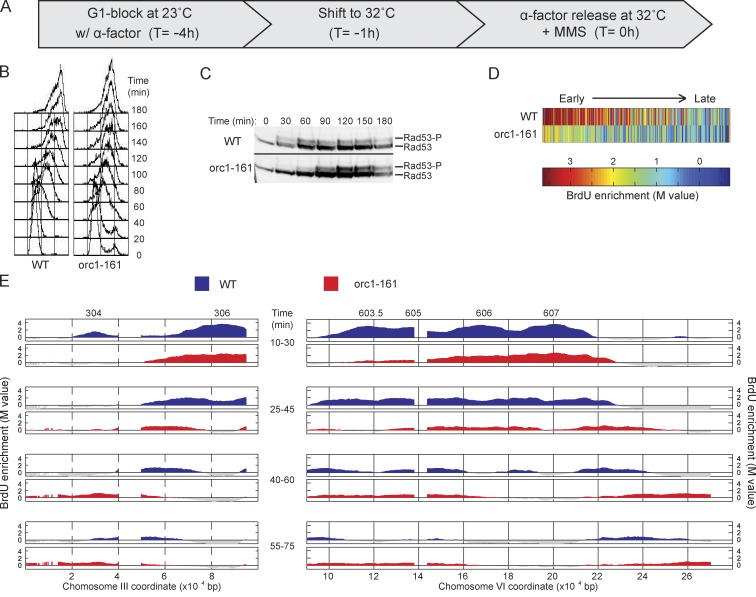
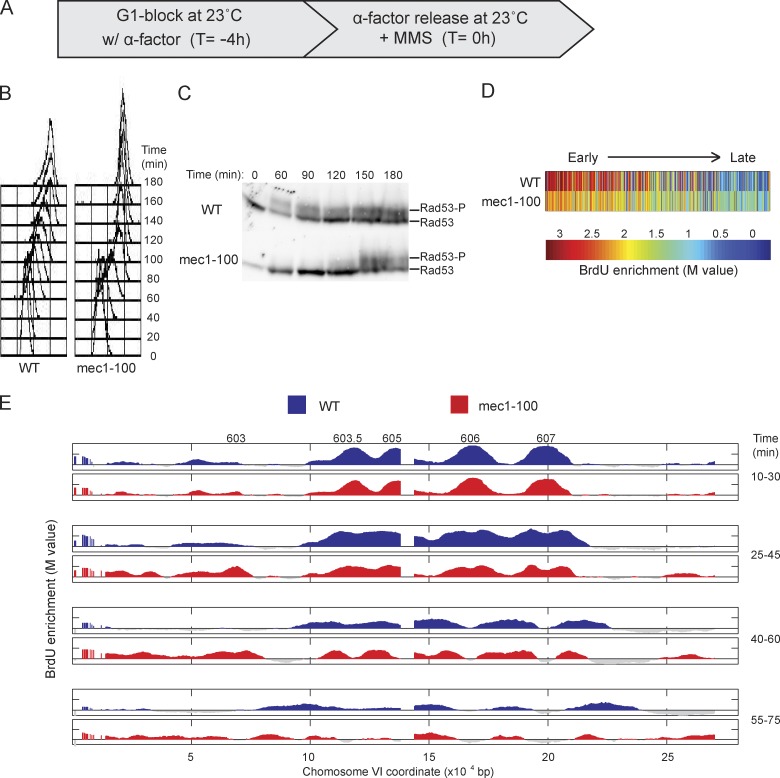
DNA damage slows DNA synthesis at replication forks; however, the mechanisms remain unclear. Cdc7 kinase is required for replication origin activation, is a target of the intra-S checkpoint, and is implicated in the response to replication fork stress. Remarkably, we found that replication forks proceed more rapidly in cells lacking Cdc7 function than in wild-type cells. We traced this effect to reduced origin firing, which results in fewer replication forks and a consequent decrease in Rad53 checkpoint signaling. Depletion of Orc1, which acts in origin firing differently than Cdc7, had similar effects as Cdc7 depletion, consistent with decreased origin firing being the source of these defects. In contrast, mec1-100 cells, which initiate excess origins and also are deficient in checkpoint activation, showed slower fork progression, suggesting the number of active forks influences their rate, perhaps as a result of competition for limiting factors.
DNA 损伤会减缓复制叉处的 DNA 合成;然而,其具体机制尚不清楚。Cdc7 激酶是复制起始激活所必需的,是细胞内 S 期检查点的靶标,并且与复制叉压力的反应有关。值得注意的是,我们发现缺乏 Cdc7 功能的细胞中复制叉的推进速度比野生型细胞更快。我们将这种效应追溯到起始点火的减少,这导致更少的复制叉,并导致 Rad53 检查点信号传导的相应减少。与 Cdc7 不同作用于起始点火的 Orc1 的耗竭具有与 Cdc7 耗竭相似的效果,这与起始点火的减少是这些缺陷的来源一致。相比之下,引发过多起始点且检查点激活也不足的 mec1-100 细胞显示出较慢的叉推进,这表明活跃叉的数量会影响其速度,这可能是由于竞争限制因素所致。