eScience Group, Microsoft Research, Los Angeles, CA 90024, United States.
Sci Rep. 2013;3:1815. doi: 10.1038/srep01815.
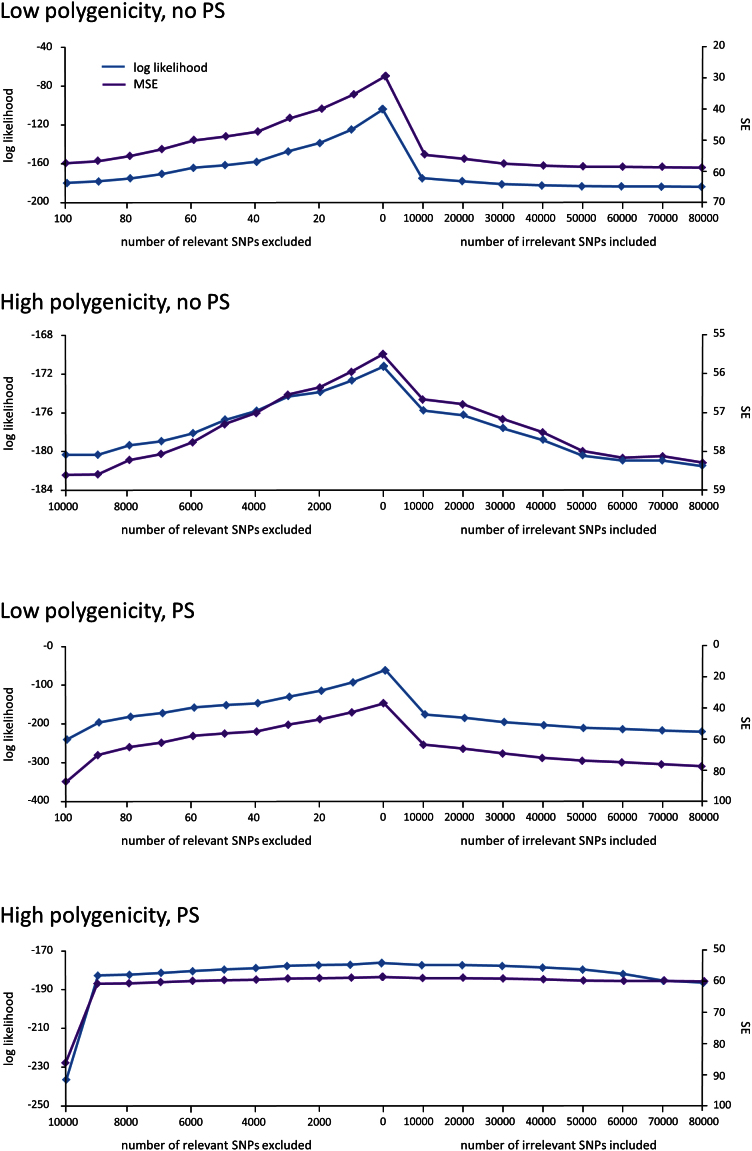
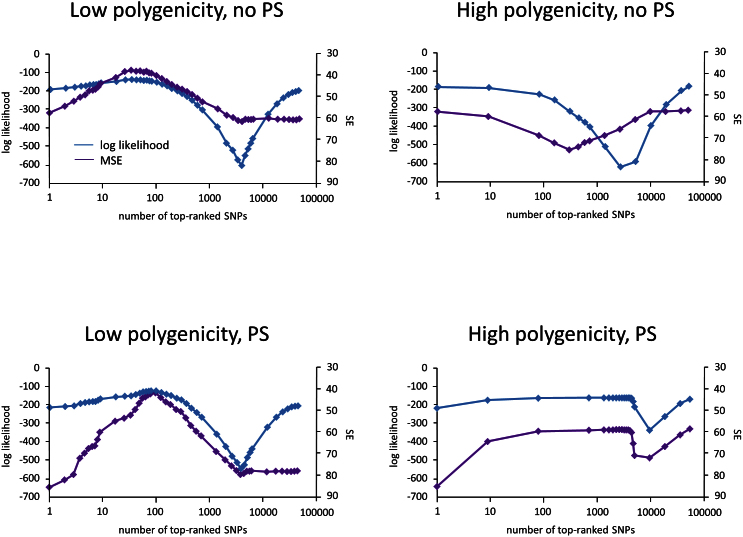
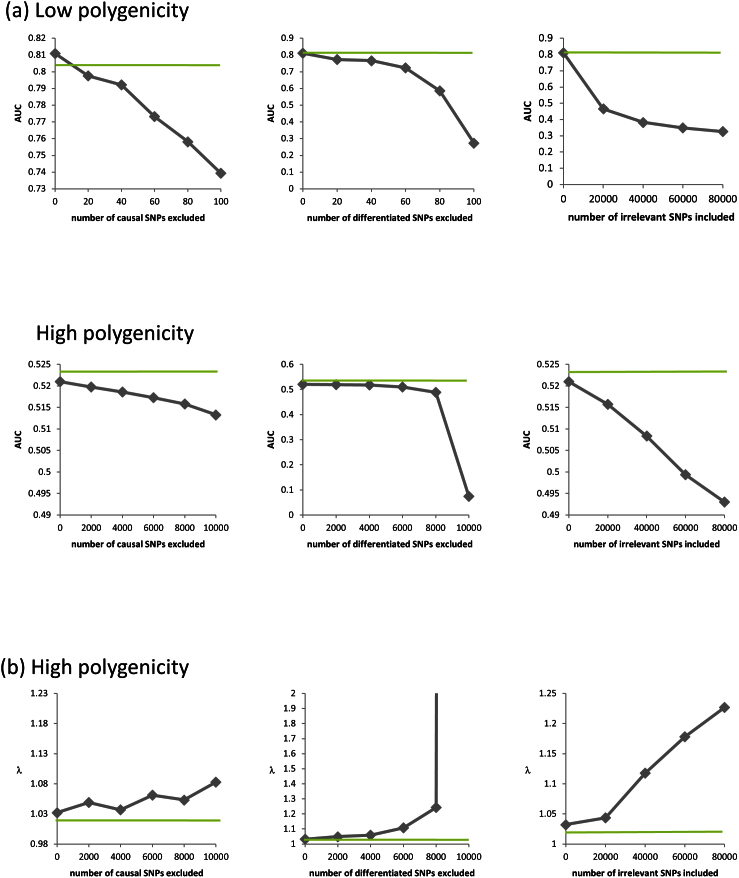
Applications of linear mixed models (LMMs) to problems in genomics include phenotype prediction, correction for confounding in genome-wide association studies, estimation of narrow sense heritability, and testing sets of variants (e.g., rare variants) for association. In each of these applications, the LMM uses a genetic similarity matrix, which encodes the pairwise similarity between every two individuals in a cohort. Although ideally these similarities would be estimated using strictly variants relevant to the given phenotype, the identity of such variants is typically unknown. Consequently, relevant variants are excluded and irrelevant variants are included, both having deleterious effects. For each application of the LMM, we review known effects and describe new effects showing how variable selection can be used to mitigate them.
线性混合模型 (LMM) 在基因组学问题中的应用包括表型预测、全基因组关联研究中的混杂因素校正、狭义遗传力估计以及变体组(例如稀有变体)的关联检验。在这些应用中,LMM 使用遗传相似性矩阵,该矩阵编码队列中每两个人之间的成对相似性。尽管理想情况下,这些相似性应该使用与给定表型严格相关的变体进行估计,但此类变体的身份通常是未知的。因此,相关变体被排除在外,无关变体被包含在内,两者都有有害影响。对于 LMM 的每种应用,我们都回顾了已知的影响,并描述了新的影响,展示了如何使用变量选择来减轻这些影响。