eScience Group, Microsoft Research, Los Angeles, CA 90024, USA.
Bioinformatics. 2013 Jun 15;29(12):1526-33. doi: 10.1093/bioinformatics/btt177. Epub 2013 Apr 18.
Approaches for testing sets of variants, such as a set of rare or common variants within a gene or pathway, for association with complex traits are important. In particular, set tests allow for aggregation of weak signal within a set, can capture interplay among variants and reduce the burden of multiple hypothesis testing. Until now, these approaches did not address confounding by family relatedness and population structure, a problem that is becoming more important as larger datasets are used to increase power.
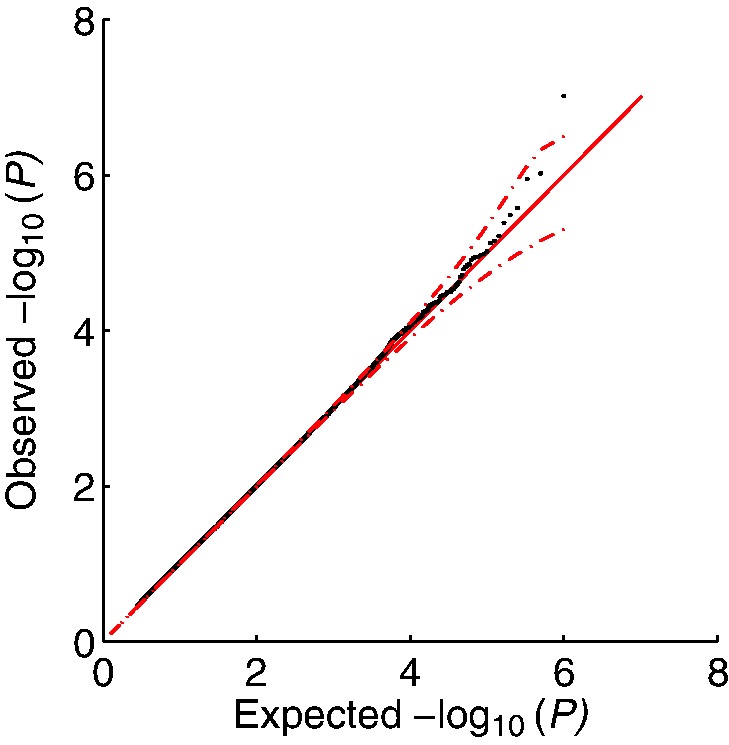
We introduce a new approach for set tests that handles confounders. Our model is based on the linear mixed model and uses two random effects-one to capture the set association signal and one to capture confounders. We also introduce a computational speedup for two random-effects models that makes this approach feasible even for extremely large cohorts. Using this model with both the likelihood ratio test and score test, we find that the former yields more power while controlling type I error. Application of our approach to richly structured Genetic Analysis Workshop 14 data demonstrates that our method successfully corrects for population structure and family relatedness, whereas application of our method to a 15 000 individual Crohn's disease case-control cohort demonstrates that it additionally recovers genes not recoverable by univariate analysis.
A Python-based library implementing our approach is available at http://mscompbio.codeplex.com.
对于测试变体集合(例如基因或途径内的一组罕见或常见变体)与复杂性状的关联的方法非常重要。特别是,集合检验允许在集合内聚集弱信号,可以捕捉变体之间的相互作用,并减少多重假设检验的负担。到目前为止,这些方法并没有解决由家族相关性和群体结构引起的混杂问题,随着使用更大的数据集来提高功效,这个问题变得越来越重要。
我们引入了一种新的集合检验方法来处理混杂因素。我们的模型基于线性混合模型,并使用两个随机效应-一个用于捕获集合关联信号,一个用于捕获混杂因素。我们还引入了一种针对两个随机效应模型的计算加速方法,即使对于非常大的队列,该方法也具有可行性。使用该模型进行似然比检验和得分检验,我们发现前者在控制 I 型错误的同时获得了更高的功效。将我们的方法应用于结构丰富的遗传分析研讨会 14 数据表明,我们的方法成功地纠正了群体结构和家族相关性,而将我们的方法应用于 15000 名个体克罗恩病病例对照队列表明,它还可以恢复单变量分析无法恢复的基因。
我们的方法的基于 Python 的库可在 http://mscompbio.codeplex.com 上获得。