Department of Food Science, Faculty of Science, University of Copenhagen, Copenhagen, Denmark.
PLoS One. 2013 May 1;8(5):e62578. doi: 10.1371/journal.pone.0062578. Print 2013.
A number of human diseases such as obesity and diabetes are associated with changes or imbalances in the gut microbiota (GM). Laboratory mice are commonly used as experimental models for such disorders. The introduction and dynamic development of next generation sequencing techniques have enabled detailed mapping of the GM of both humans and animal models. Nevertheless there is still a significant knowledge gap regarding the human and mouse common GM core and thus the applicability of the latter as an animal model. The aim of the present study was to identify inter- and intra-individual differences and similarities between the GM composition of particular mouse strains and humans.
METHODOLOGY/PRINCIPAL FINDINGS: A total of 1509428 high quality tag-encoded partial 16S rRNA gene sequences determined using 454/FLX Titanium (Roche) pyro-sequencing reflecting the GM composition of 32 human samples from 16 individuals and 88 mouse samples from three laboratory mouse strains commonly used in diabetes research were analyzed using Principal Coordinate Analysis (PCoA), nonparametric multivariate analysis of similarity (ANOSIM) and alpha diversity measures. A reliable cutoff threshold for low abundant taxa estimated on the basis of the present study is recommended for similar trials.
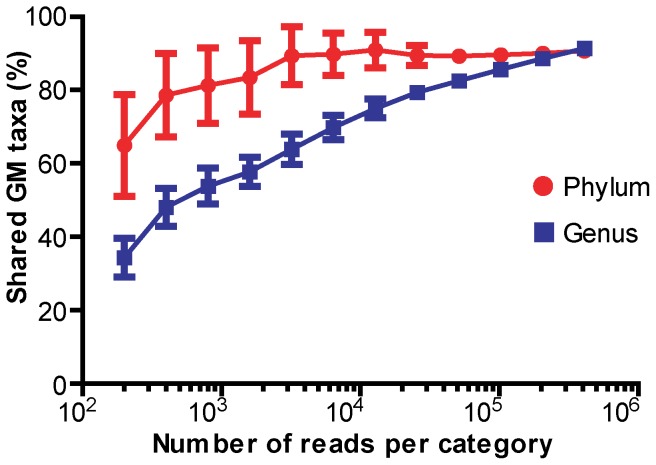
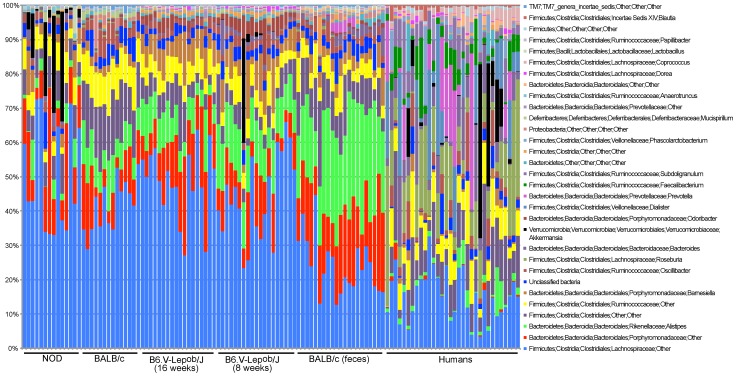
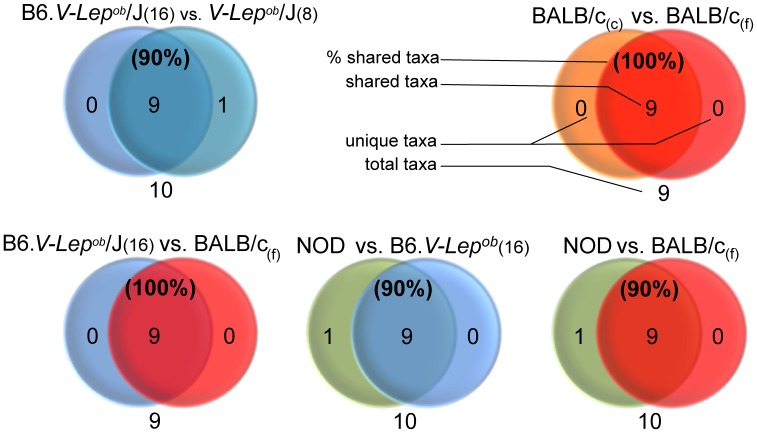
CONCLUSIONS/SIGNIFICANCE: Distinctive quantitative differences in the relative abundance of most taxonomic groups between the examined categories were found. All investigated mouse strains clustered separately, but with a range of shared features when compared to the human GM. However, both mouse fecal, caecal and human fecal samples shared to a large extent not only representatives of the same phyla, but also a substantial fraction of common genera, where the number of shared genera increased with sequencing depth. In conclusion, the GM of mice and humans is quantitatively different (in terms of abundance of specific phyla and species) but share a large qualitatively similar core.
许多人类疾病,如肥胖症和糖尿病,与肠道微生物群(GM)的变化或失衡有关。实验小鼠通常被用作此类疾病的实验模型。下一代测序技术的引入和动态发展使人类和动物模型的 GM 图谱得以详细绘制。然而,关于人类和小鼠共同 GM 核心以及后者作为动物模型的适用性,仍然存在着显著的知识差距。本研究的目的是确定特定小鼠品系和人类 GM 组成之间的个体间和个体内差异和相似性。
方法/主要发现:使用 454/FLX Titanium(Roche)焦磷酸测序技术,共测定了 1509428 个高质量标记编码的部分 16S rRNA 基因序列,反映了 32 个来自 16 个个体的人类样本和 3 个常用于糖尿病研究的实验室小鼠品系的 88 个小鼠样本的 GM 组成。使用主坐标分析(PCoA)、非参数多元相似性分析(ANOSIM)和 alpha 多样性测度对这些数据进行了分析。本研究建议为类似试验推荐一个可靠的低丰度分类单元的截断阈值。
结论/意义:在所检查的类别中,大多数分类群的相对丰度存在明显的定量差异。所有研究的小鼠品系均单独聚类,但与人类 GM 相比具有一系列共同特征。然而,小鼠粪便、盲肠和人类粪便样本不仅在同一门的代表方面有很大程度的共享,而且还共享了大量的共同属,随着测序深度的增加,共同属的数量也在增加。总之,小鼠和人类的 GM 在数量上是不同的(特定门和物种的丰度),但具有很大的相似核心。