Key Laboratory of Marine Genetics and Breeding (MGB), Ministry of Education, College of Marine Life Sciences, Ocean University of China, Qingdao, China.
PLoS One. 2013 May 7;8(5):e63927. doi: 10.1371/journal.pone.0063927. Print 2013.
Bivalves play an important role in the ecosystems they inhabit and represent an important food source all over the world. So far limited genetic research has focused on this group of animals largely due to the lack of sufficient genetic or genomic resources. Here, we performed de novo transcriptome sequencing to produce the most comprehensive expressed sequence tag resource for Zhikong scallop (Chlamys farreri), and conducted the first transcriptome comparison for scallops.
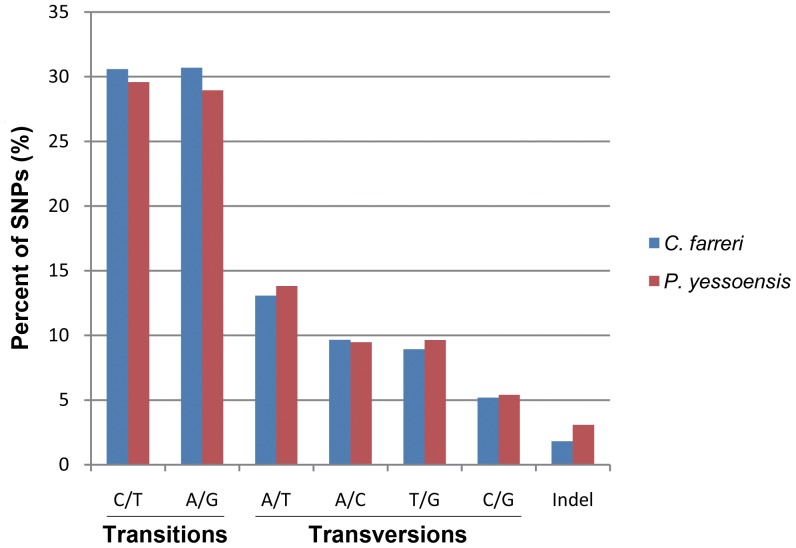
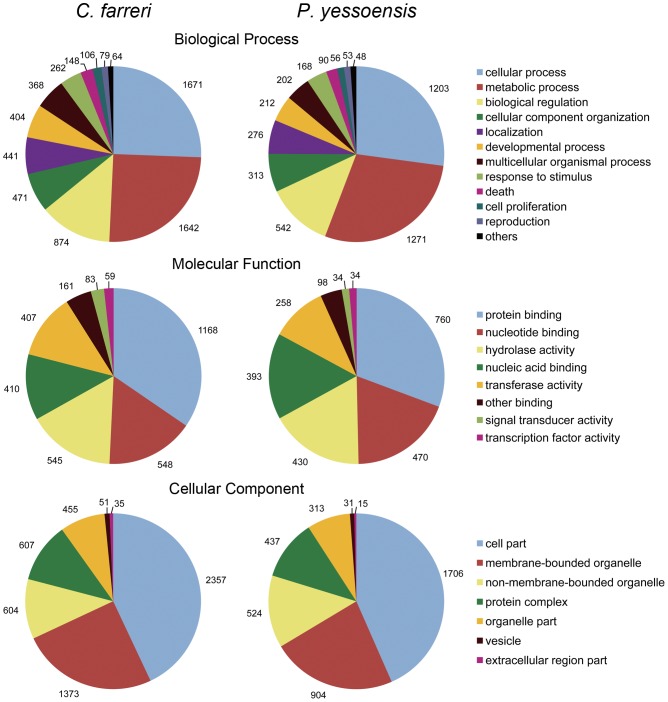
In a single 454 sequencing run, 1,033,636 reads were produced and then assembled into 26,165 contigs. These contigs were then clustered into 24,437 isotigs and further grouped into 20,056 isogroups. About 47% of the isogroups showed significant matches to known proteins based on sequence similarity. Transcripts putatively involved in growth, reproduction and stress/immune-response were identified through Gene ontology (GO) and KEGG pathway analyses. Transcriptome comparison with Yesso scallop (Patinopecten yessoensis) revealed similar patterns of GO representation. Moreover, 38 putative fast-evolving genes were identified through analyzing the orthologous gene pairs between the two scallop species. More than 46,000 single nucleotide polymorphisms (SNPs) and 350 simple sequence repeats (SSRs) were also detected.
Our study provides the most comprehensive transcriptomic resource currently available for C. farreri. Based on this resource, we performed the first large-scale transcriptome comparison between the two scallop species, C. farreri and P. yessoensis, and identified a number of putative fast-evolving genes, which may play an important role in scallop speciation and/or local adaptation. A large set of single nucleotide polymorphisms and simple sequence repeats were identified, which are ready for downstream marker development. This transcriptomic resource should lay an important foundation for future genetic or genomic studies on C. farreri.
双壳类在其栖息的生态系统中发挥着重要作用,是世界各地的重要食物来源。迄今为止,由于缺乏足够的遗传或基因组资源,对该类动物的遗传研究非常有限。在这里,我们进行了从头转录组测序,为栉孔扇贝(Chlamys farreri)生成了最全面的表达序列标签资源,并进行了首次扇贝转录组比较。
在单个 454 测序运行中,生成了 1,033,636 条读取序列,然后将其组装成 26,165 条 contigs。这些 contigs 随后聚类成 24,437 条 isotigs,并进一步分为 20,056 条 isogroups。根据序列相似性,约 47%的 isogroups与已知蛋白质具有显著匹配。通过基因本体(GO)和 KEGG 通路分析鉴定了推定参与生长、繁殖和应激/免疫反应的转录本。与虾夷扇贝(Patinopecten yessoensis)的转录组比较显示出相似的 GO 表达模式。此外,通过分析两个扇贝物种的同源基因对,鉴定了 38 个推定快速进化的基因。还检测到超过 46,000 个单核苷酸多态性(SNP)和 350 个简单序列重复(SSR)。
我们的研究为 C. farreri 提供了目前最全面的转录组资源。基于该资源,我们对两个扇贝物种 C. farreri 和 P. yessoensis 进行了首次大规模转录组比较,并鉴定了一些推定快速进化的基因,这些基因可能在扇贝物种形成和/或局部适应中发挥重要作用。鉴定出了大量的单核苷酸多态性和简单序列重复,可用于下游标记的开发。这个转录组资源应该为未来对 C. farreri 的遗传或基因组研究奠定重要基础。