Xu Dong, Zhang Yang
Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI 48109, USA.
Sci Rep. 2013;3:1895. doi: 10.1038/srep01895.
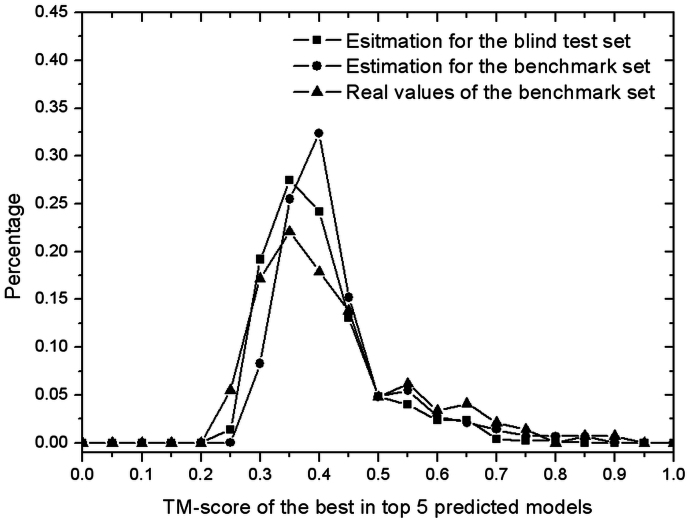
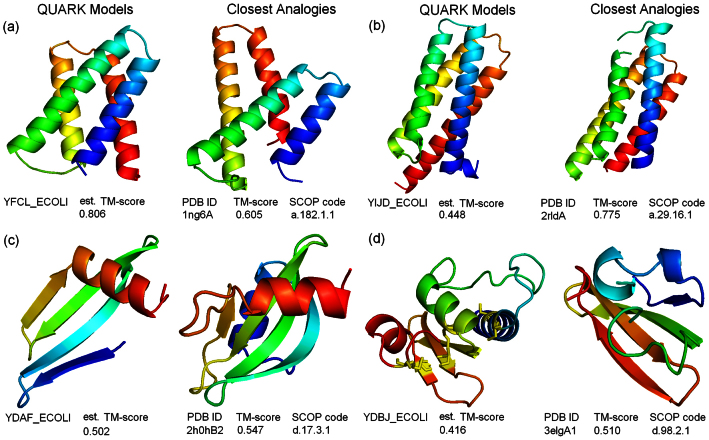
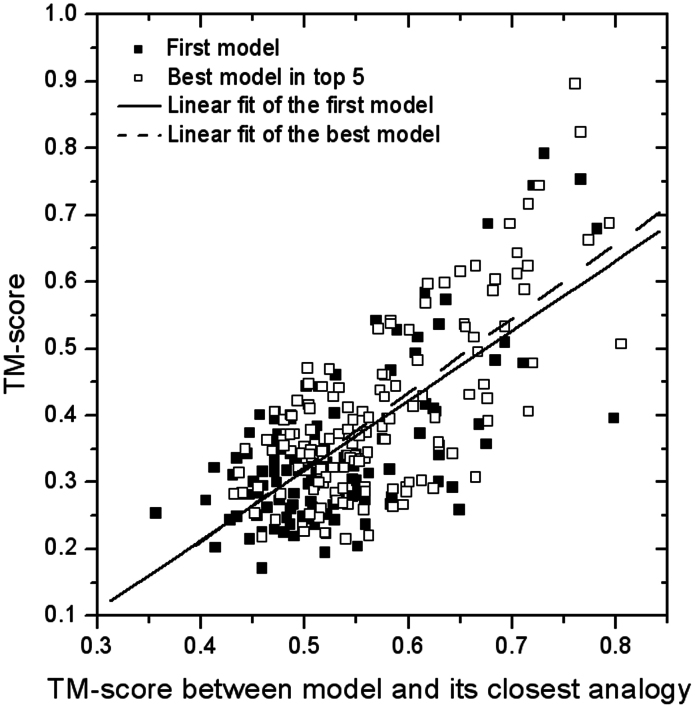
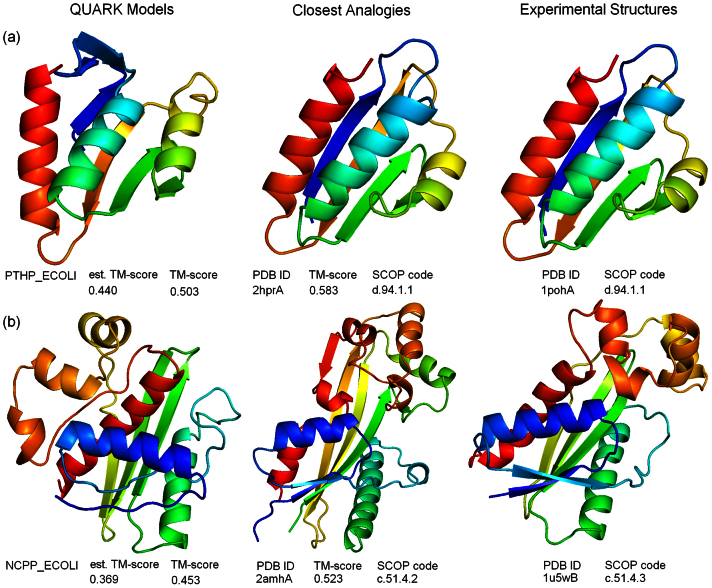
Genome-wide protein structure prediction and structure-based function annotation have been a long-term goal in molecular biology but not yet become possible due to difficulties in modeling distant-homology targets. We developed a hybrid pipeline combining ab initio folding and template-based modeling for genome-wide structure prediction applied to the Escherichia coli genome. The pipeline was tested on 43 known sequences, where QUARK-based ab initio folding simulation generated models with TM-score 17% higher than that by traditional comparative modeling methods. For 495 unknown hard sequences, 72 are predicted to have a correct fold (TM-score > 0.5) and 321 have a substantial portion of structure correctly modeled (TM-score > 0.35). 317 sequences can be reliably assigned to a SCOP fold family based on structural analogy to existing proteins in PDB. The presented results, as a case study of E. coli, represent promising progress towards genome-wide structure modeling and fold family assignment using state-of-the-art ab initio folding algorithms.
全基因组蛋白质结构预测和基于结构的功能注释一直是分子生物学的长期目标,但由于对远源同源靶点进行建模存在困难,目前尚未实现。我们开发了一种混合流程,将从头折叠和基于模板的建模相结合,用于全基因组结构预测,并应用于大肠杆菌基因组。该流程在43个已知序列上进行了测试,基于QUARK的从头折叠模拟生成的模型,其TM分数比传统比较建模方法高出17%。对于495个未知的难处理序列,预计有72个具有正确的折叠(TM分数>0.5),321个的大部分结构被正确建模(TM分数>0.35)。基于与PDB中现有蛋白质的结构相似性,317个序列可以可靠地归类到一个SCOP折叠家族。作为大肠杆菌的案例研究,所展示的结果代表了在使用最先进的从头折叠算法进行全基因组结构建模和折叠家族归类方面取得的有前景的进展。