Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA.
Department of Biological Chemistry, University of Michigan, Ann Arbor, MI, USA.
Nat Commun. 2021 Aug 18;12(1):5011. doi: 10.1038/s41467-021-25316-w.
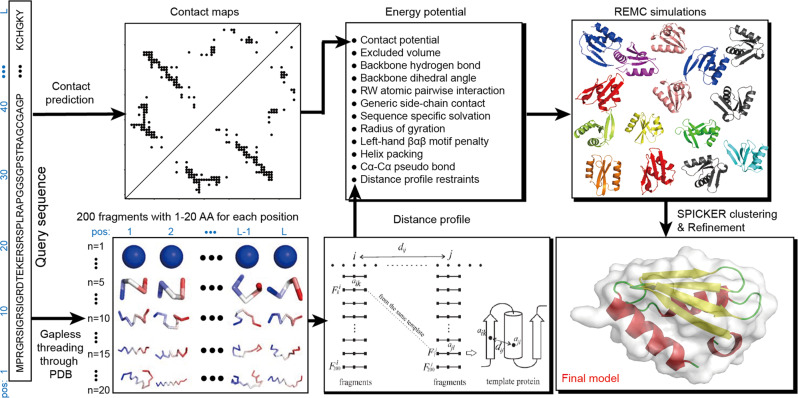
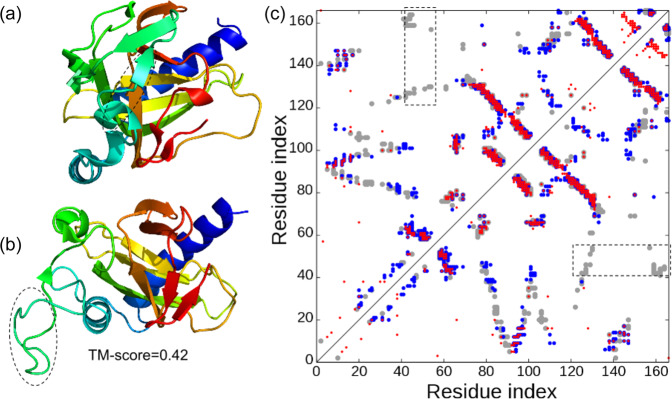
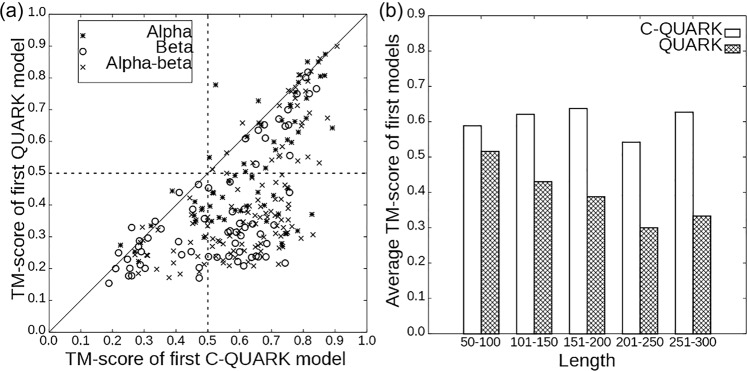
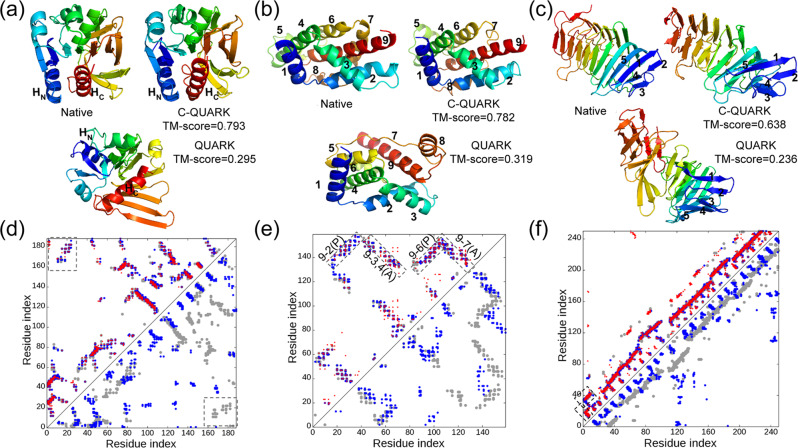
Sequence-based contact prediction has shown considerable promise in assisting non-homologous structure modeling, but it often requires many homologous sequences and a sufficient number of correct contacts to achieve correct folds. Here, we developed a method, C-QUARK, that integrates multiple deep-learning and coevolution-based contact-maps to guide the replica-exchange Monte Carlo fragment assembly simulations. The method was tested on 247 non-redundant proteins, where C-QUARK could fold 75% of the cases with TM-scores (template-modeling scores) ≥0.5, which was 2.6 times more than that achieved by QUARK. For the 59 cases that had either low contact accuracy or few homologous sequences, C-QUARK correctly folded 6 times more proteins than other contact-based folding methods. C-QUARK was also tested on 64 free-modeling targets from the 13th CASP (critical assessment of protein structure prediction) experiment and had an average GDT_TS (global distance test) score that was 5% higher than the best CASP predictors. These data demonstrate, in a robust manner, the progress in modeling non-homologous protein structures using low-accuracy and sparse contact-map predictions.
基于序列的接触预测在辅助非同源结构建模方面显示出了很大的潜力,但它通常需要许多同源序列和足够数量的正确接触来实现正确的折叠。在这里,我们开发了一种方法 C-QUARK,它集成了多种基于深度学习和共进化的接触图,以指导 replica-exchange Monte Carlo 片段组装模拟。该方法在 247 个非冗余蛋白质上进行了测试,C-QUARK 可以折叠 75%的 TM-scores(模板建模分数)≥0.5 的情况,比 QUARK 高 2.6 倍。对于接触准确性低或同源序列较少的 59 个案例,C-QUARK 正确折叠的蛋白质比其他基于接触的折叠方法多 6 倍。C-QUARK 还在第 13 届 CASP(蛋白质结构预测关键评估)实验的 64 个自由建模靶标上进行了测试,其平均 GDT_TS(全局距离测试)得分比最佳的 CASP 预测器高 5%。这些数据以稳健的方式证明了使用低准确性和稀疏接触图预测来建模非同源蛋白质结构的进展。