Department of Computer Science, University of California-Irvine, CA, USA.
BMC Bioinformatics. 2013;14 Suppl 5(Suppl 5):S11. doi: 10.1186/1471-2105-14-S5-S11. Epub 2013 Apr 10.
RNA-seq, a next-generation sequencing based method for transcriptome analysis, is rapidly emerging as the method of choice for comprehensive transcript abundance estimation. The accuracy of RNA-seq can be highly impacted by the purity of samples. A prominent, outstanding problem in RNA-seq is how to estimate transcript abundances in heterogeneous tissues, where a sample is composed of more than one cell type and the inhomogeneity can substantially confound the transcript abundance estimation of each individual cell type. Although experimental methods have been proposed to dissect multiple distinct cell types, computationally "deconvoluting" heterogeneous tissues provides an attractive alternative, since it keeps the tissue sample as well as the subsequent molecular content yield intact.
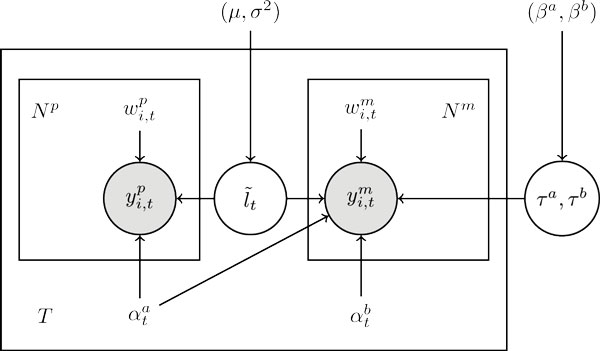
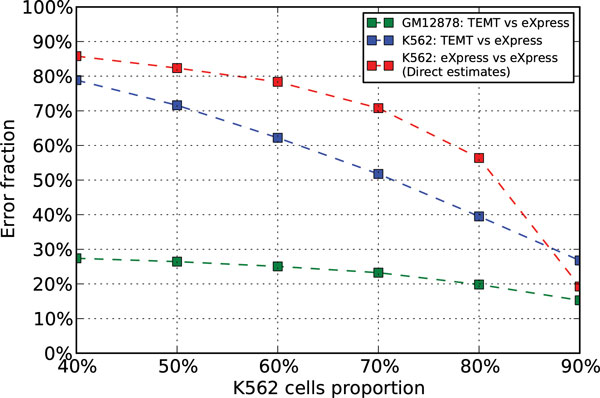
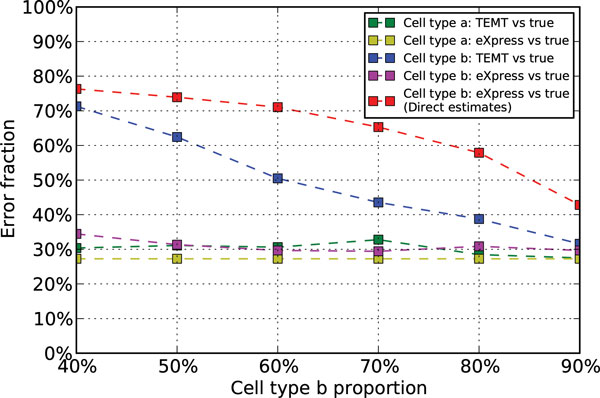
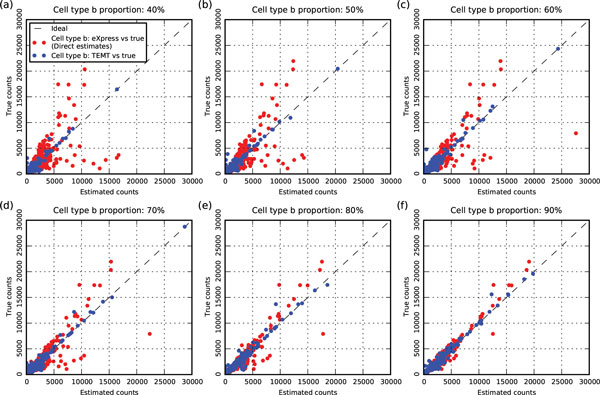
Here we propose a probabilistic model-based approach, Transcript Estimation from Mixed Tissue samples (TEMT), to estimate the transcript abundances of each cell type of interest from RNA-seq data of heterogeneous tissue samples. TEMT incorporates positional and sequence-specific biases, and its online EM algorithm only requires a runtime proportional to the data size and a small constant memory. We test the proposed method on both simulation data and recently released ENCODE data, and show that TEMT significantly outperforms current state-of-the-art methods that do not take tissue heterogeneity into account. Currently, TEMT only resolves the tissue heterogeneity resulting from two cell types, but it can be extended to handle tissue heterogeneity resulting from multi cell types. TEMT is written in python, and is freely available at https://github.com/uci-cbcl/TEMT.
The probabilistic model-based approach proposed here provides a new method for analyzing RNA-seq data from heterogeneous tissue samples. By applying the method to both simulation data and ENCODE data, we show that explicitly accounting for tissue heterogeneity can significantly improve the accuracy of transcript abundance estimation.
RNA-seq 是一种基于下一代测序的转录组分析方法,正在迅速成为全面转录丰度估计的首选方法。RNA-seq 的准确性可能会受到样品纯度的高度影响。RNA-seq 的一个突出问题是如何估计异质组织中的转录本丰度,其中一个样品由超过一种细胞类型组成,不均匀性会严重干扰每个单个细胞类型的转录本丰度估计。尽管已经提出了实验方法来分离多个不同的细胞类型,但通过计算“解卷积”异质组织提供了一种有吸引力的替代方法,因为它保持了组织样品以及随后的分子含量的完整性。
在这里,我们提出了一种基于概率模型的方法,即混合组织样本的转录本估计(TEMT),从异质组织样本的 RNA-seq 数据中估计每个感兴趣的细胞类型的转录本丰度。TEMT 结合了位置和序列特异性偏差,其在线 EM 算法仅需要与数据大小成比例的运行时间和少量的常数内存。我们在模拟数据和最近发布的 ENCODE 数据上测试了所提出的方法,并表明 TEMT 显著优于当前不考虑组织异质性的最先进方法。目前,TEMT 仅解决由两种细胞类型引起的组织异质性,但它可以扩展到处理由多种细胞类型引起的组织异质性。TEMT 是用 python 编写的,可在 https://github.com/uci-cbcl/TEMT 上免费获得。
这里提出的基于概率模型的方法为分析异质组织样本的 RNA-seq 数据提供了一种新方法。通过将该方法应用于模拟数据和 ENCODE 数据,我们表明明确考虑组织异质性可以显著提高转录本丰度估计的准确性。