ETH Zürich, Department of Chemistry and Applied Biosciences, Institute of Pharmaceutical Sciences, Zürich, Switzerland.
PLoS Comput Biol. 2013;9(6):e1003088. doi: 10.1371/journal.pcbi.1003088. Epub 2013 Jun 6.
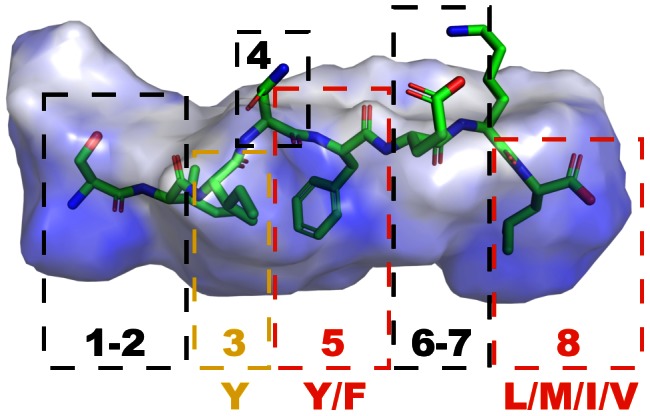
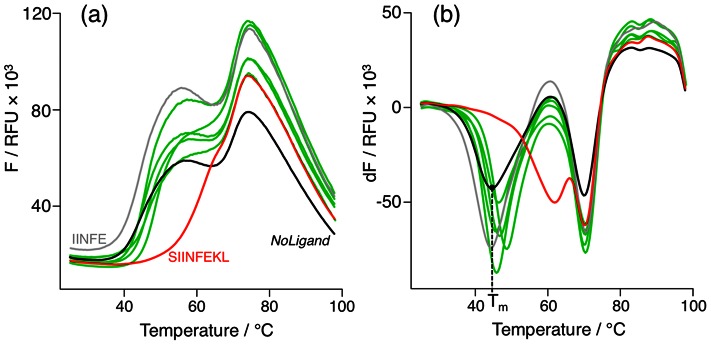
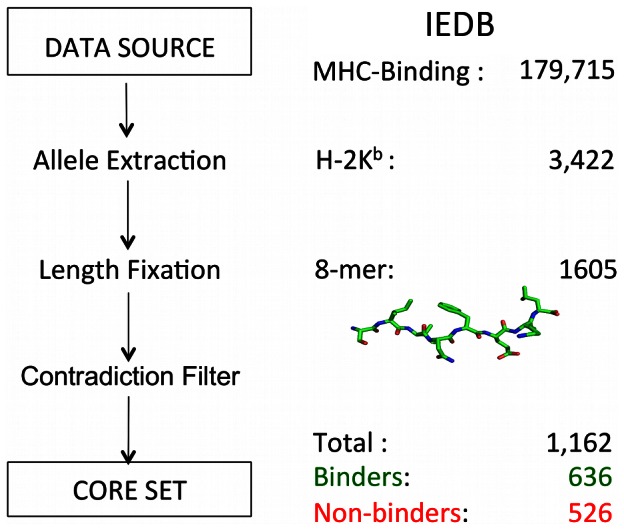
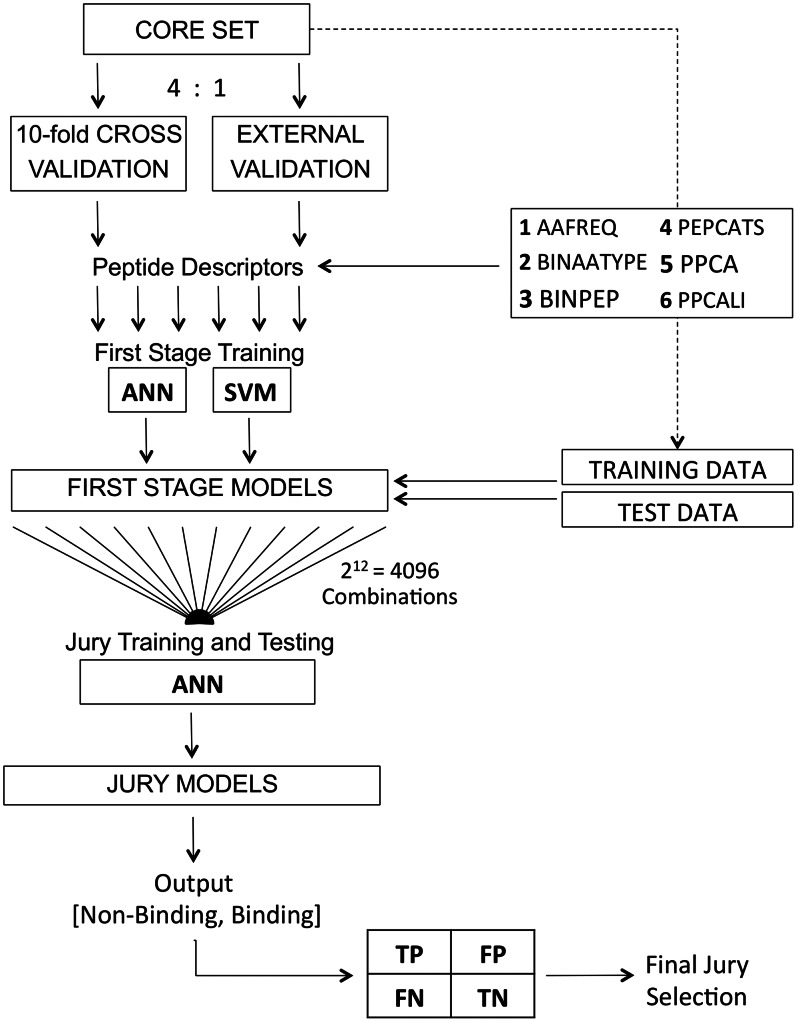
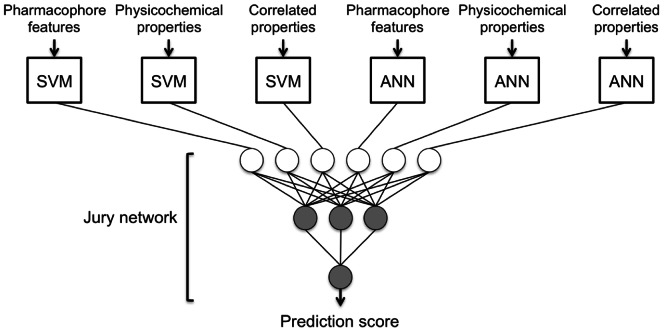
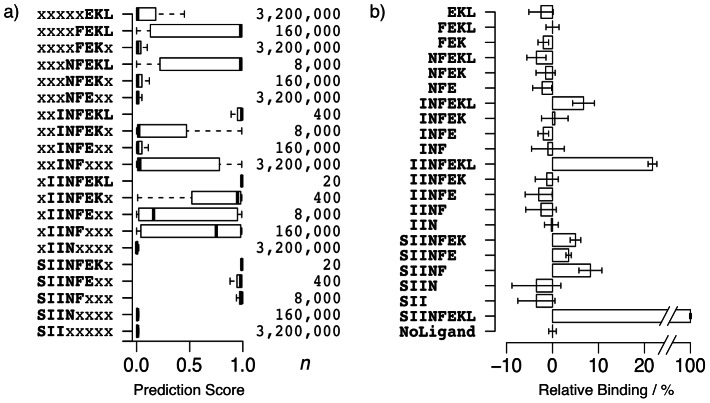
Designed peptides that bind to major histocompatibility protein I (MHC-I) allomorphs bear the promise of representing epitopes that stimulate a desired immune response. A rigorous bioinformatical exploration of sequence patterns hidden in peptides that bind to the mouse MHC-I allomorph H-2K(b) is presented. We exemplify and validate these motif findings by systematically dissecting the epitope SIINFEKL and analyzing the resulting fragments for their binding potential to H-2K(b) in a thermal denaturation assay. The results demonstrate that only fragments exclusively retaining the carboxy- or amino-terminus of the reference peptide exhibit significant binding potential, with the N-terminal pentapeptide SIINF as shortest ligand. This study demonstrates that sophisticated machine-learning algorithms excel at extracting fine-grained patterns from peptide sequence data and predicting MHC-I binding peptides, thereby considerably extending existing linear prediction models and providing a fresh view on the computer-based molecular design of future synthetic vaccines. The server for prediction is available at http://modlab-cadd.ethz.ch (SLiDER tool, MHC-I version 2012).
设计与主要组织相容性蛋白 I (MHC-I) 同种异型结合的肽有望代表刺激所需免疫反应的表位。本文对与小鼠 MHC-I 同种异型 H-2K(b) 结合的肽中隐藏的序列模式进行了严格的生物信息学探索。我们通过系统地剖析表位 SIINFEKL 并分析所得片段在热变性测定中与 H-2K(b) 的结合潜力,对这些基序发现进行了例证和验证。结果表明,只有完全保留参考肽的羧基或氨基末端的片段表现出显著的结合潜力,最短的配体为 N 末端五肽 SIINF。这项研究表明,复杂的机器学习算法擅长从肽序列数据中提取细微的模式并预测 MHC-I 结合肽,从而大大扩展了现有的线性预测模型,并为未来合成疫苗的计算机分子设计提供了新的视角。预测服务器可在 http://modlab-cadd.ethz.ch 上获得(SLiDER 工具,MHC-I 版本 2012)。