Department of Genetics, Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, Missouri 63108, USA.
Genome Res. 2013 Sep;23(9):1541-53. doi: 10.1101/gr.152231.112. Epub 2013 Jun 26.
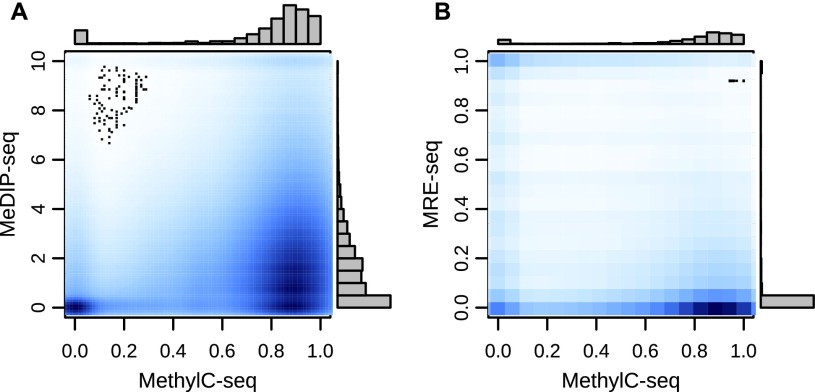
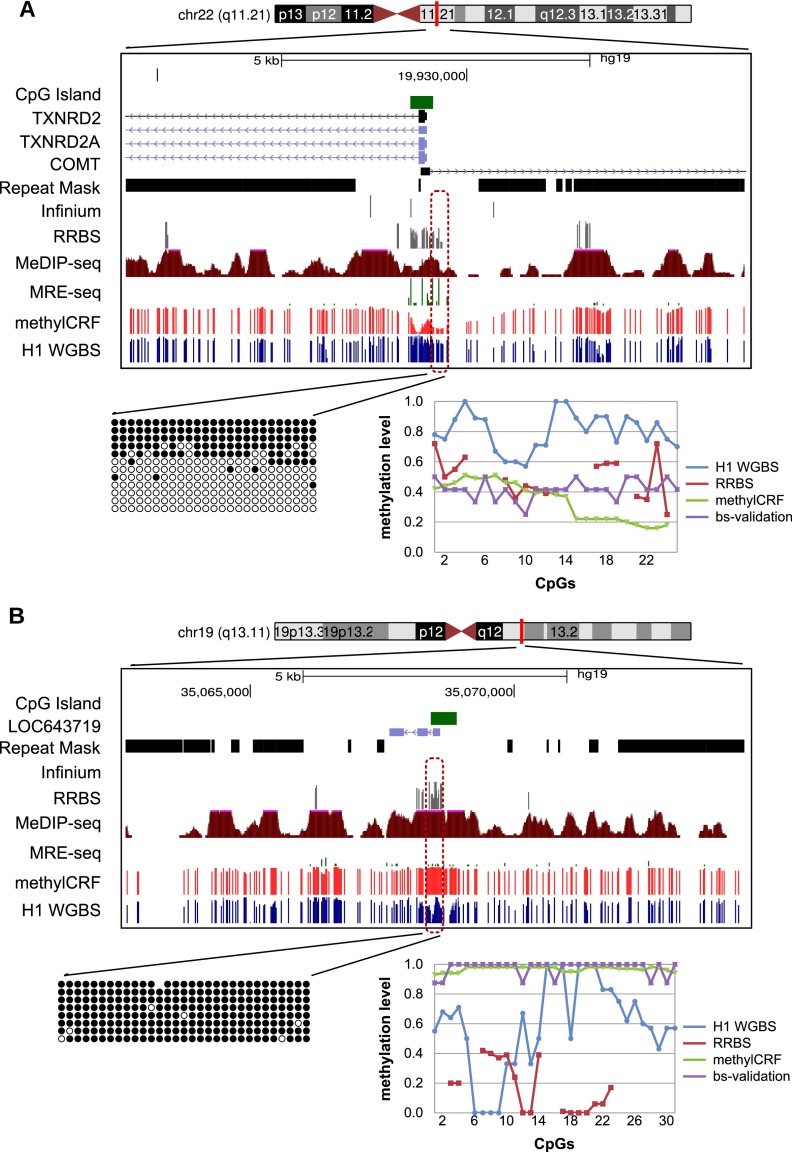
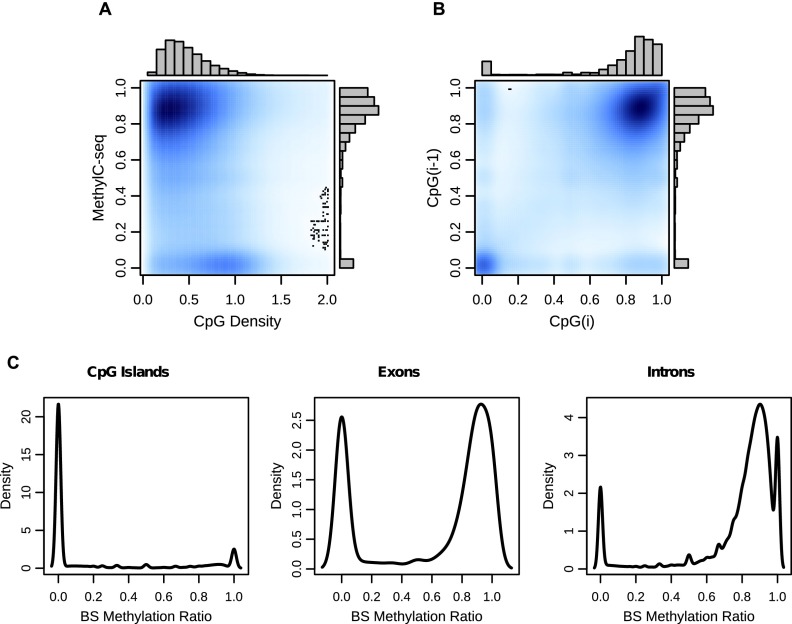
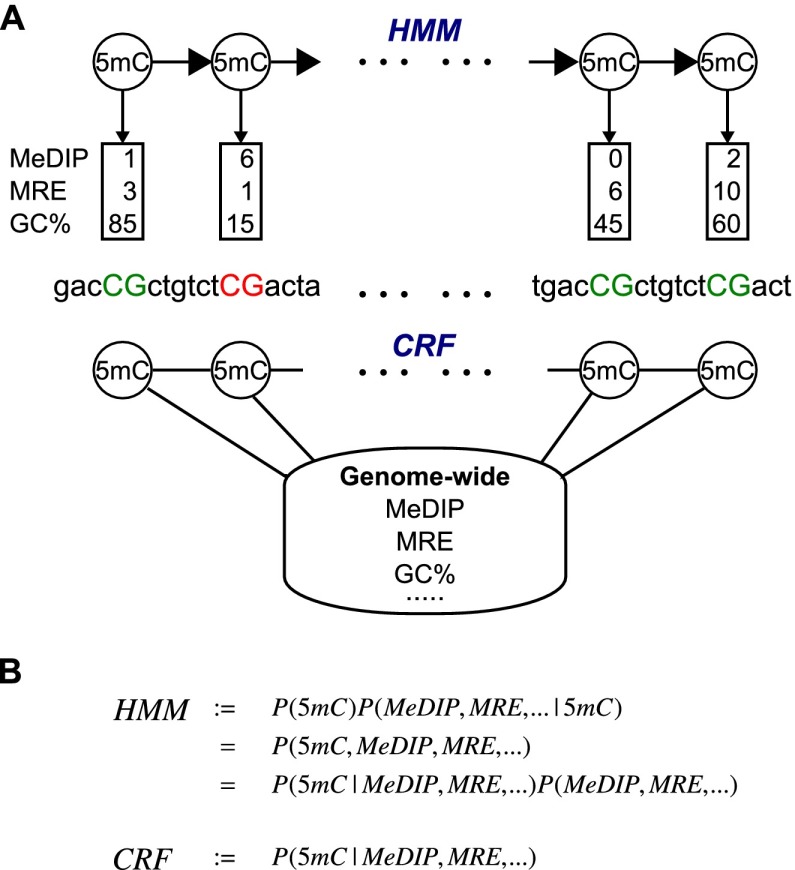
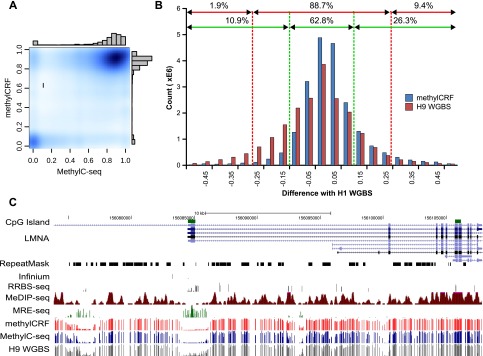
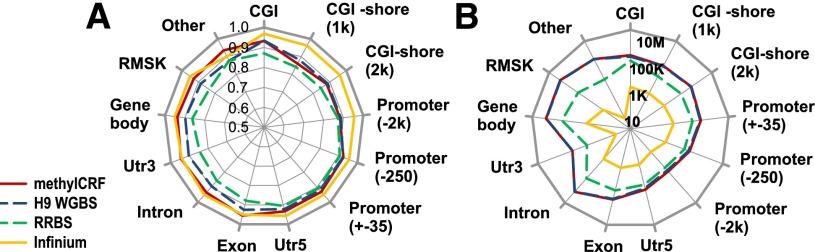
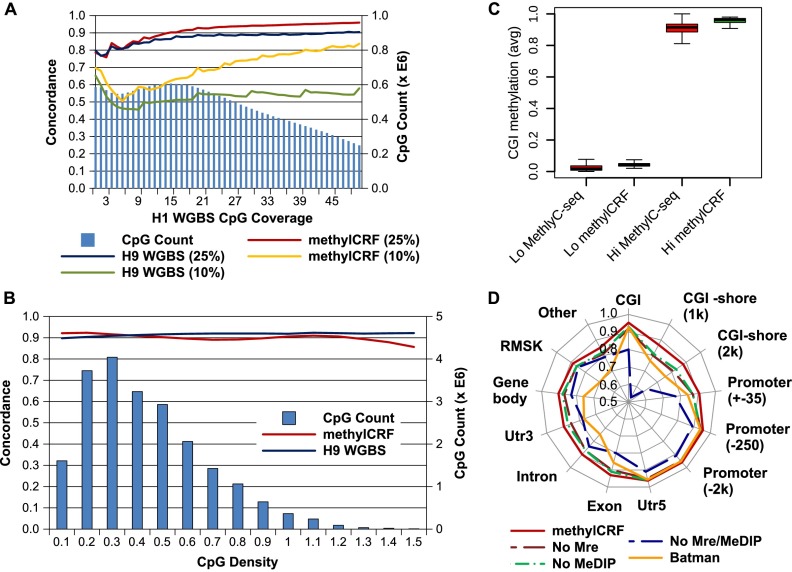
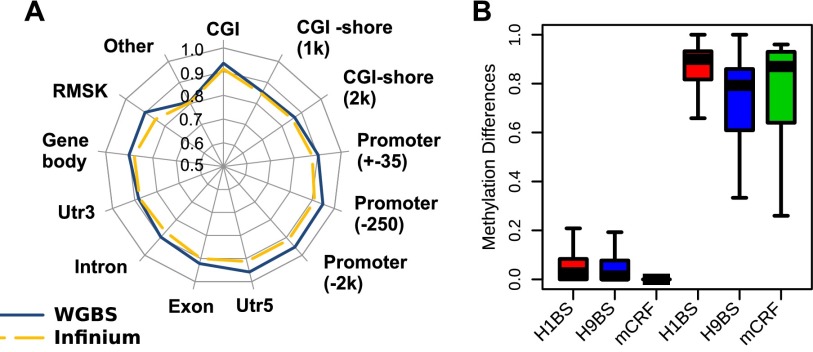
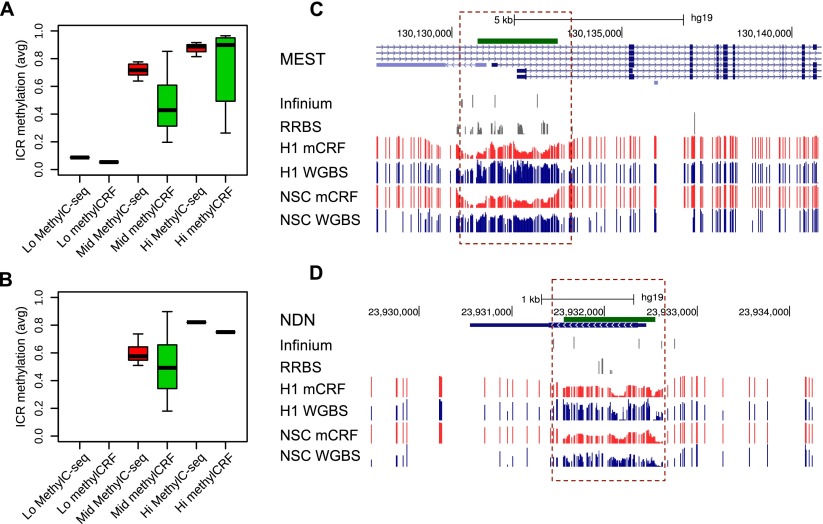
Recent advancements in sequencing-based DNA methylation profiling methods provide an unprecedented opportunity to map complete DNA methylomes. These include whole-genome bisulfite sequencing (WGBS, MethylC-seq, or BS-seq), reduced-representation bisulfite sequencing (RRBS), and enrichment-based methods such as MeDIP-seq, MBD-seq, and MRE-seq. These methods yield largely comparable results but differ significantly in extent of genomic CpG coverage, resolution, quantitative accuracy, and cost, at least while using current algorithms to interrogate the data. None of these existing methods provides single-CpG resolution, comprehensive genome-wide coverage, and cost feasibility for a typical laboratory. We introduce methylCRF, a novel conditional random fields-based algorithm that integrates methylated DNA immunoprecipitation (MeDIP-seq) and methylation-sensitive restriction enzyme (MRE-seq) sequencing data to predict DNA methylation levels at single-CpG resolution. Our method is a combined computational and experimental strategy to produce DNA methylomes of all 28 million CpGs in the human genome for a fraction (<10%) of the cost of whole-genome bisulfite sequencing methods. methylCRF was benchmarked for accuracy against Infinium arrays, RRBS, WGBS sequencing, and locus-specific bisulfite sequencing performed on the same human embryonic stem cell line. methylCRF transformation of MeDIP-seq/MRE-seq was equivalent to a biological replicate of WGBS in quantification, coverage, and resolution. We used conventional bisulfite conversion, PCR, cloning, and sequencing to validate loci where our predictions do not agree with whole-genome bisulfite data, and in 11 out of 12 cases, methylCRF predictions of methylation level agree better with validated results than does whole-genome bisulfite sequencing. Therefore, methylCRF transformation of MeDIP-seq/MRE-seq data provides an accurate, inexpensive, and widely accessible strategy to create full DNA methylomes.
基于测序的 DNA 甲基化分析方法的最新进展为绘制完整的 DNA 甲基化组图谱提供了前所未有的机会。这些方法包括全基因组亚硫酸氢盐测序(WGBS、MethylC-seq 或 BS-seq)、简化代表性亚硫酸氢盐测序(RRBS)以及基于富集的方法,如 MeDIP-seq、MBD-seq 和 MRE-seq。这些方法的结果大致相当,但在基因组 CpG 覆盖范围、分辨率、定量准确性和成本方面存在显著差异,至少在使用当前算法来分析数据时是如此。这些现有的方法都无法提供单个 CpG 的分辨率、全面的全基因组覆盖范围以及对典型实验室来说具有成本效益的可行性。我们介绍了 methylCRF,这是一种基于条件随机场的新型算法,它整合了甲基化 DNA 免疫沉淀(MeDIP-seq)和甲基敏感限制性内切酶(MRE-seq)测序数据,以预测单个 CpG 分辨率的 DNA 甲基化水平。我们的方法是一种结合计算和实验的策略,可以以全基因组亚硫酸氢盐测序方法成本的一小部分(<10%)生成人类基因组中所有 2800 万个 CpG 的 DNA 甲基化组。methylCRF 在准确性方面与 Infinium 微阵列、RRBS、WGBS 测序和在相同的人类胚胎干细胞系上进行的特定基因座的亚硫酸氢盐测序进行了基准测试。methylCRF 对 MeDIP-seq/MRE-seq 的转化在定量、覆盖范围和分辨率方面与 WGBS 的生物学重复相当。我们使用常规的亚硫酸氢盐转化、PCR、克隆和测序来验证我们的预测与全基因组亚硫酸氢盐数据不一致的基因座,在 12 个基因座中的 11 个基因座中,methylCRF 对甲基化水平的预测比全基因组亚硫酸氢盐测序更符合验证结果。因此,MeDIP-seq/MRE-seq 数据的 methylCRF 转化为创建完整的 DNA 甲基化组图谱提供了一种准确、经济且广泛适用的策略。