Department of Obstetrics and Gynecology, Beijing Aviation General Hospital, Beijing, 100012, China.
Department of Stomatology, Chinese PLA General Hospital, Beijing, 100853, People's Republic of China.
BMC Biotechnol. 2018 Feb 6;18(1):7. doi: 10.1186/s12896-017-0409-7.
Detection of DNA methylome at single-base resolution is a significant challenge but promises to shed considerable light on human disease etiology. Current technologies could not detect DNA methylation genome-wide at single-base resolution with small amount of sequencing data and could not avoid detecting the methylation of repetitive elements which are considered as "junk DNA".
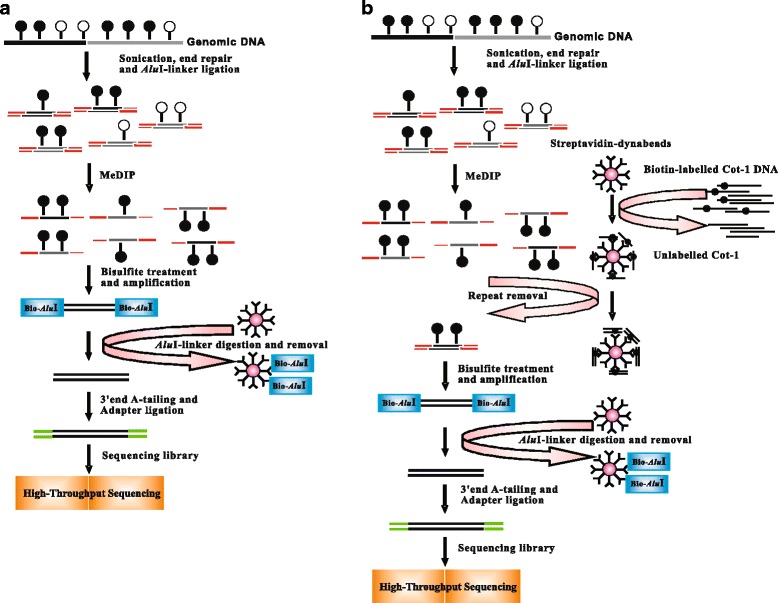
In this study, we have developed a novel DNA methylome profiling technology named MB-seq with its ability to identify genome-wide 5mC and quantify DNA methylation levels by introduced an assistant adapter AluI-linker This linker can be ligated to sonicated DNA and then be digested after the bisulfite treatment and amplification, which has no effect of MeDIP enrichment. Because many researchers are interested in investigating the methylation of functional regions such as promoters and gene bodies, we have also developed a novel alternative method named MRB-seq, which can be used to investigate the DNA methylation of functional regions by removing the repeats with Cot-1 DNA.
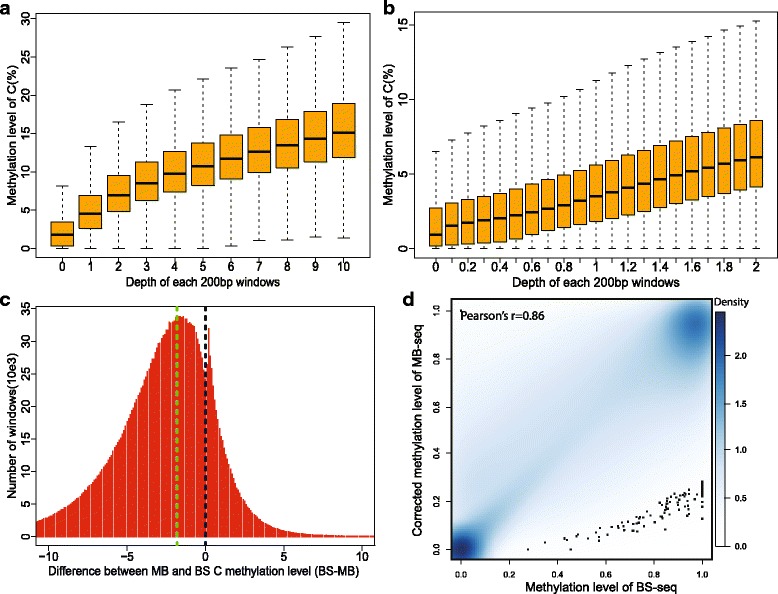
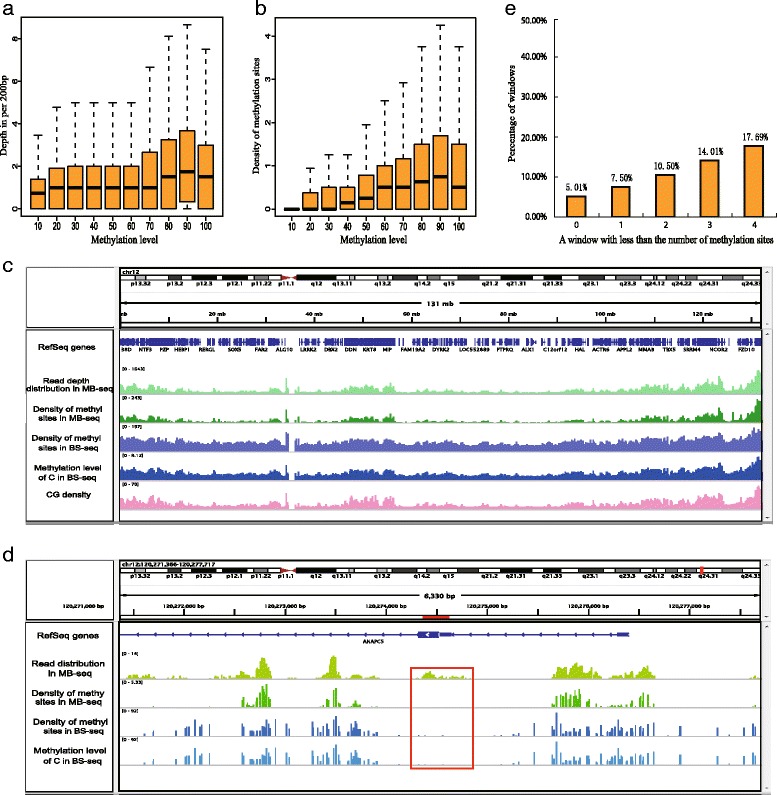
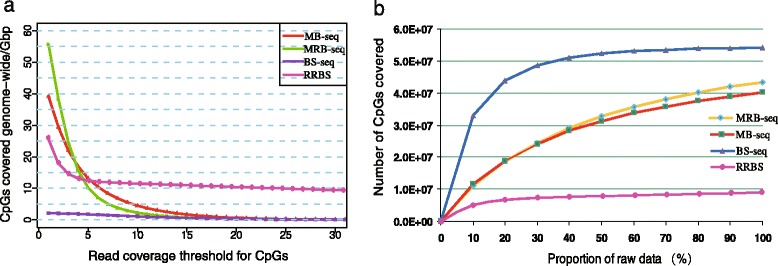
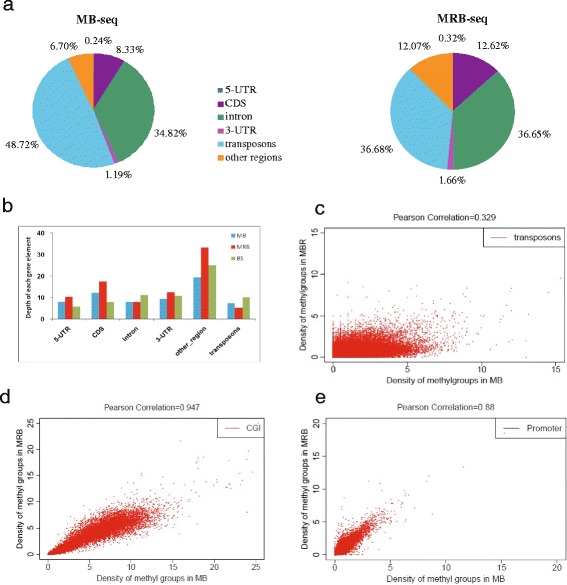
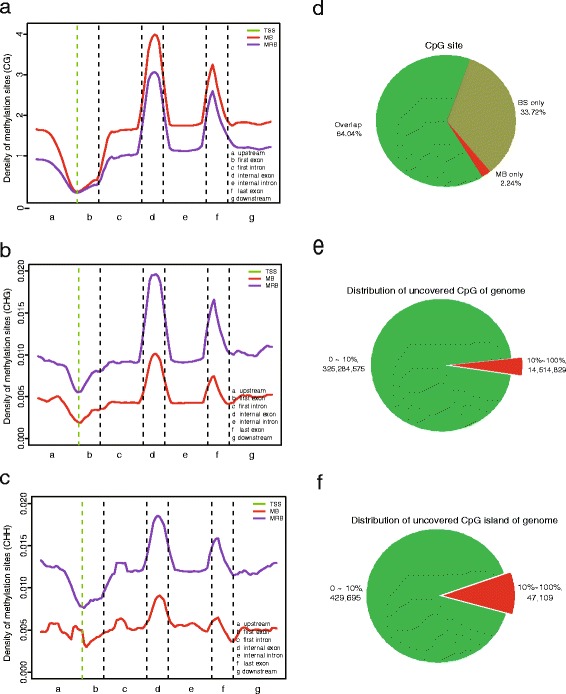
In this study, we have developed MB-seq, a novel DNA methylome profiling technology combining MeDIP-seq with bisulfite conversion, which can precisely detect the 5mC sites and determine their DNA methylation level at single-base resolution in a cost-effective way. In addition, we have developed a new alternative method, MRB-seq (MeDIP-repetitive elements removal-bisulfite sequencing), which interrogates 5mCs in functional regions by depleting nearly half of repeat fragments enriched by MeDIP. Comparing MB-seq and MRB-seq to whole-genome BS-seq using the same batch of DNA from YH peripheral blood mononuclear cells. We found that the sequencing data of MB-seq and MRB-seq almost reaches saturation after generating 7-8 Gbp data, whereas BS-seq requires about 100 Gbp data to achieve the same effect. In comparison to MeDIP-seq and BS-seq, MB-seq offers several key advantages, including single-base resolution, discriminating the methylated sites within a CpG and non-CpG pattern and overcoming the false positive of MeDIP-seq due to the non-specific binding of 5-methylcytidine antibody to genomic fragments.
Our novel developed method MB-seq can accelerate the decoding process of DNA methylation mechanism in human diseases because it requires 7-8 Gbp data to measure human methylome with enough coverage and sequencing depth, affording it a direct and practical application in the study of multiple samples. In addition, we have also provided a novel alternative MRB-seq method, which removes most repetitive sequences and allows researchers to genome-wide characterize DNA methylation of functional regions.
以单碱基分辨率检测 DNA 甲基化组是一项重大挑战,但有望极大地阐明人类疾病的病因。目前的技术无法在小量测序数据的情况下以单碱基分辨率检测全基因组的 DNA 甲基化,并且无法避免检测到被认为是“垃圾 DNA”的重复元件的甲基化。
在这项研究中,我们开发了一种名为 MB-seq 的新型 DNA 甲基化组分析技术,该技术通过引入辅助接头 AluI-linker 来识别全基因组的 5mC 并定量 DNA 甲基化水平。该接头可以连接到超声处理的 DNA 上,然后在亚硫酸氢盐处理和扩增后进行消化,这不会影响 MeDIP 富集。因为许多研究人员都有兴趣研究启动子和基因体等功能区域的甲基化,所以我们还开发了一种新的替代方法,称为 MRB-seq,它可以通过去除 Cot-1 DNA 来去除重复序列,从而用于研究功能区域的 DNA 甲基化。
在这项研究中,我们开发了 MB-seq,这是一种将 MeDIP-seq 与亚硫酸氢盐转化相结合的新型 DNA 甲基化组分析技术,能够以经济有效的方式精确检测 5mC 位点并确定其单碱基分辨率的 DNA 甲基化水平。此外,我们开发了一种新的替代方法,MRB-seq(MeDIP-重复元件去除-亚硫酸氢盐测序),通过耗尽 MeDIP 富集的近一半重复片段来检测功能区域中的 5mC。使用来自 YH 外周血单核细胞的相同批次 DNA,我们将 MB-seq 和 MRB-seq 与全基因组 BS-seq 进行了比较。我们发现,MB-seq 和 MRB-seq 的测序数据在生成 7-8 Gbp 数据后几乎达到饱和,而 BS-seq 需要大约 100 Gbp 数据才能达到相同的效果。与 MeDIP-seq 和 BS-seq 相比,MB-seq 具有几个关键优势,包括单碱基分辨率、区分 CpG 和非 CpG 模式中的甲基化位点以及克服由于 5-甲基胞嘧啶抗体与基因组片段的非特异性结合而导致的 MeDIP-seq 的假阳性。
我们新开发的 MB-seq 方法可以加速人类疾病中 DNA 甲基化机制的解码过程,因为它只需要 7-8 Gbp 数据即可测量人类甲基组,具有足够的覆盖度和测序深度,因此可以直接应用于多个样本的研究。此外,我们还提供了一种新的替代方法 MRB-seq,它可以去除大部分重复序列,允许研究人员对功能区域的 DNA 甲基化进行全基因组特征分析。