Toyota Technological Institute at Chicago, 6045 S Kenwood, IL 60637, USA.
Bioinformatics. 2013 Jul 1;29(13):i266-73. doi: 10.1093/bioinformatics/btt211.
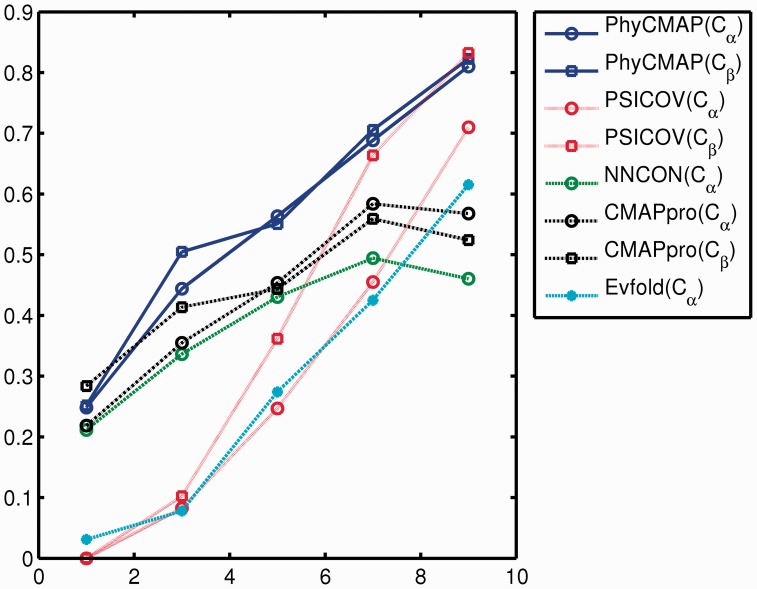
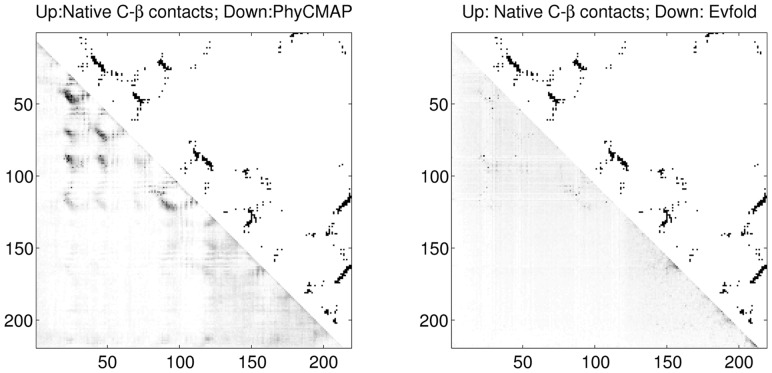
Protein contact map describes the pairwise spatial and functional relationship of residues in a protein and contains key information for protein 3D structure prediction. Although studied extensively, it remains challenging to predict contact map using only sequence information. Most existing methods predict the contact map matrix element-by-element, ignoring correlation among contacts and physical feasibility of the whole-contact map. A couple of recent methods predict contact map by using mutual information, taking into consideration contact correlation and enforcing a sparsity restraint, but these methods demand for a very large number of sequence homologs for the protein under consideration and the resultant contact map may be still physically infeasible.
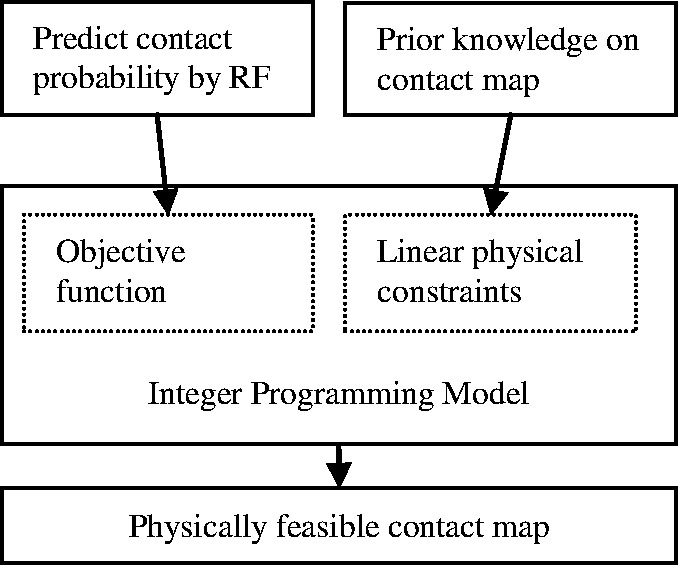
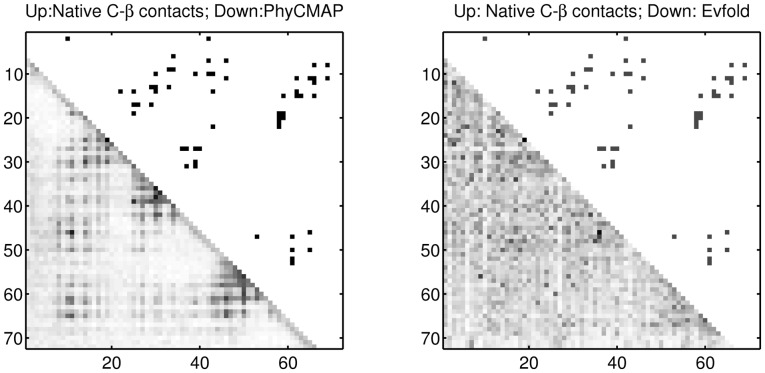
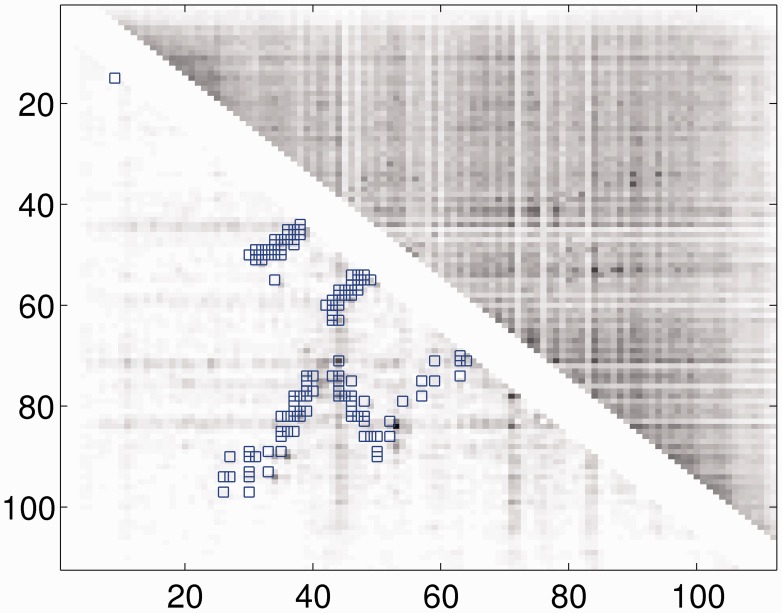
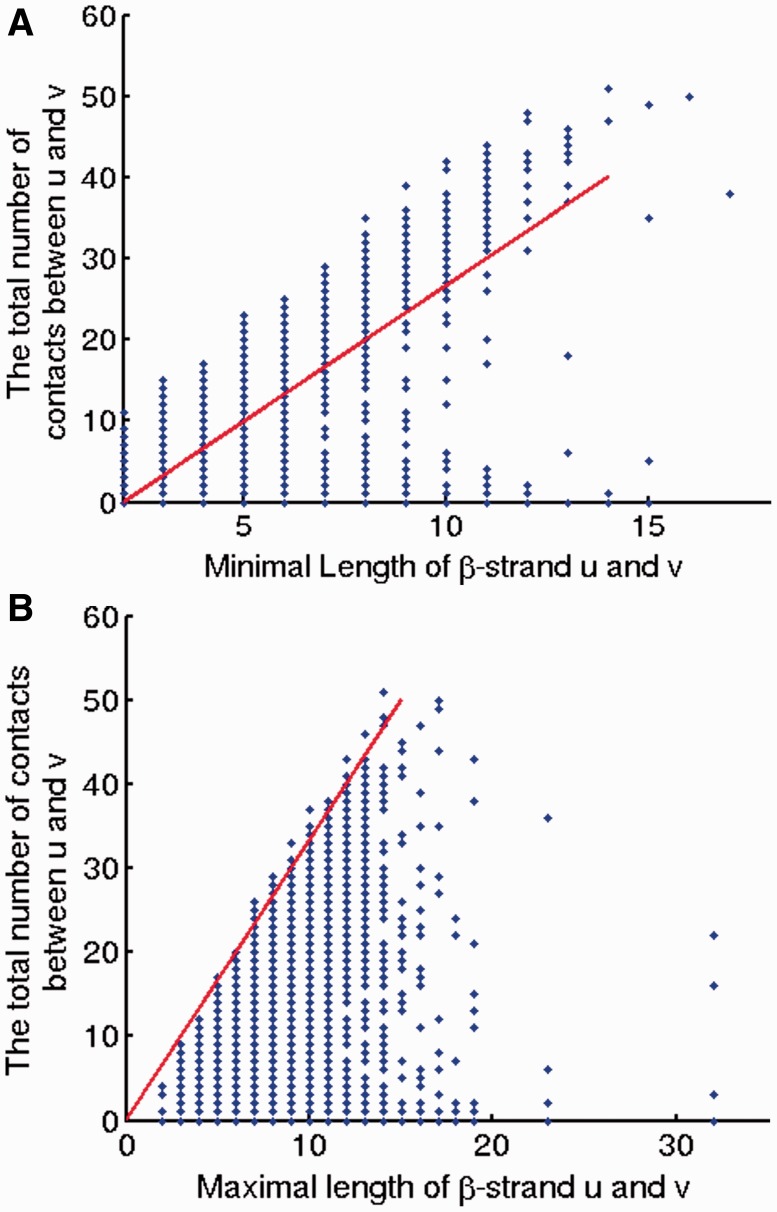
This article presents a novel method PhyCMAP for contact map prediction, integrating both evolutionary and physical restraints by machine learning and integer linear programming. The evolutionary restraints are much more informative than mutual information, and the physical restraints specify more concrete relationship among contacts than the sparsity restraint. As such, our method greatly reduces the solution space of the contact map matrix and, thus, significantly improves prediction accuracy. Experimental results confirm that PhyCMAP outperforms currently popular methods no matter how many sequence homologs are available for the protein under consideration.
蛋白质接触图描述了蛋白质中残基的成对空间和功能关系,包含了蛋白质 3D 结构预测的关键信息。尽管已经进行了广泛的研究,但仅使用序列信息预测接触图仍然具有挑战性。大多数现有的方法逐个预测接触图矩阵元素,忽略了接触之间的相关性和整个接触图的物理可行性。最近的一些方法通过使用互信息来预测接触图,考虑了接触相关性并施加了稀疏性约束,但这些方法需要大量蛋白质同源序列,并且得到的接触图可能仍然不符合物理要求。
本文提出了一种新的接触图预测方法 PhyCMAP,通过机器学习和整数线性规划整合了进化和物理约束。进化约束比互信息更具信息量,物理约束比稀疏性约束更具体地指定了接触之间的关系。因此,我们的方法大大缩小了接触图矩阵的解空间,从而显著提高了预测准确性。实验结果证实,无论对于所考虑的蛋白质有多少个序列同源物可用,PhyCMAP 都优于当前流行的方法。