YouKaryote Genomics, London, Ontario, Canada.
PLoS One. 2013 Jul 2;8(7):e67019. doi: 10.1371/journal.pone.0067019. Print 2013.
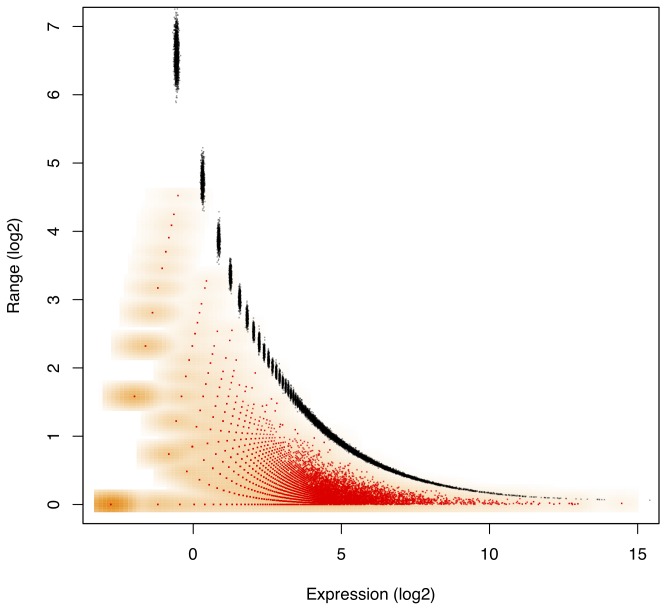
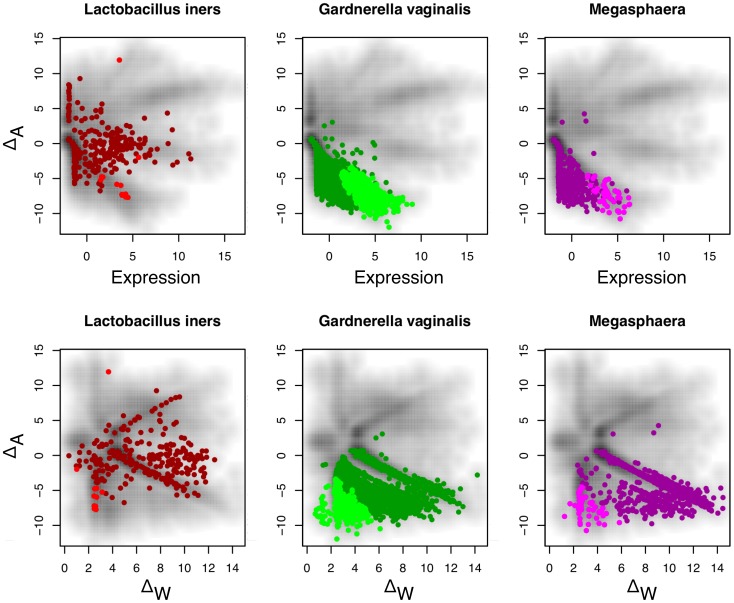
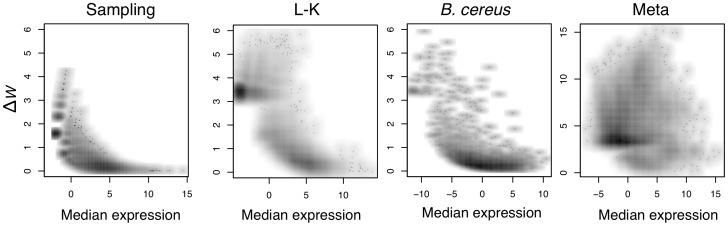
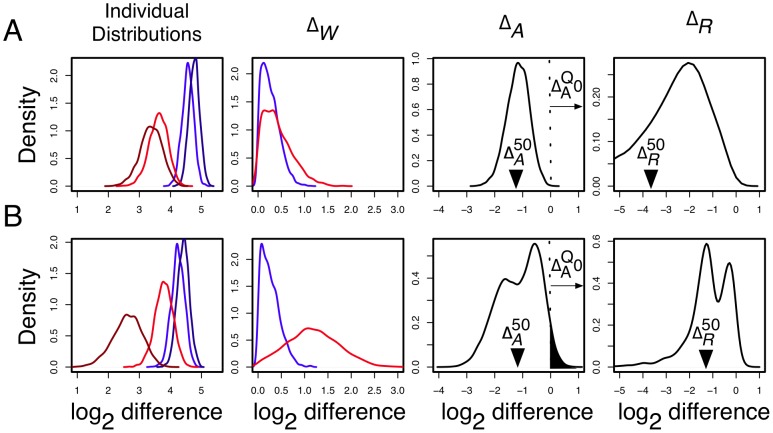
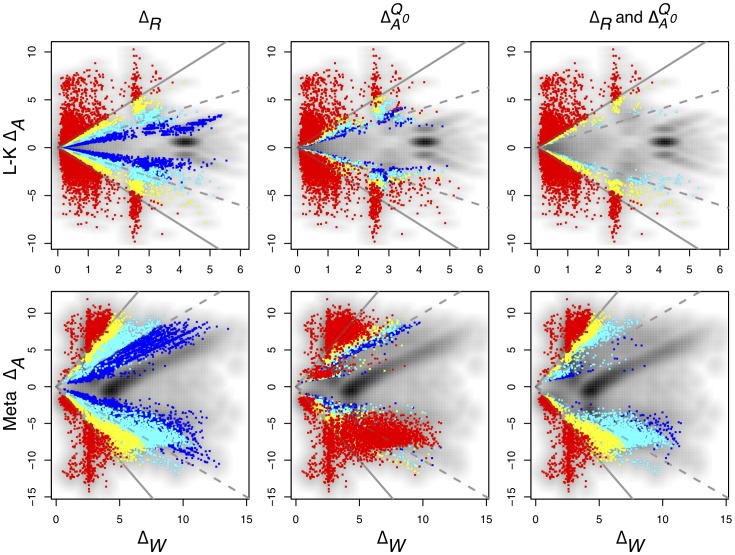
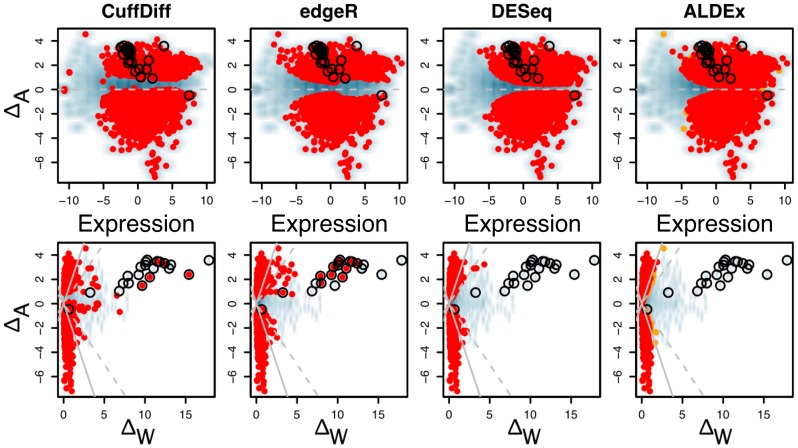
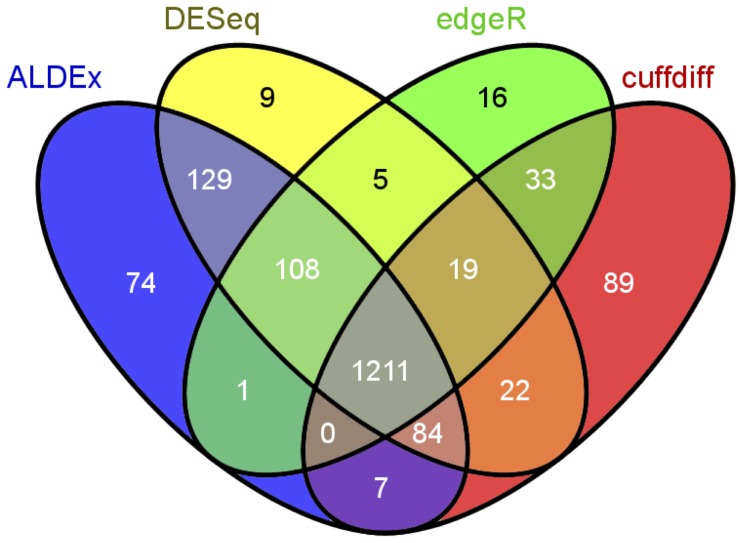
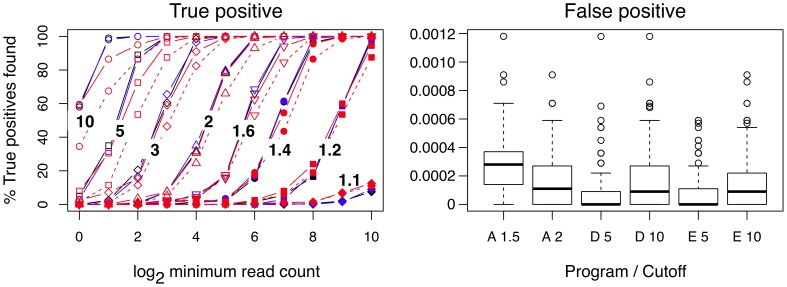
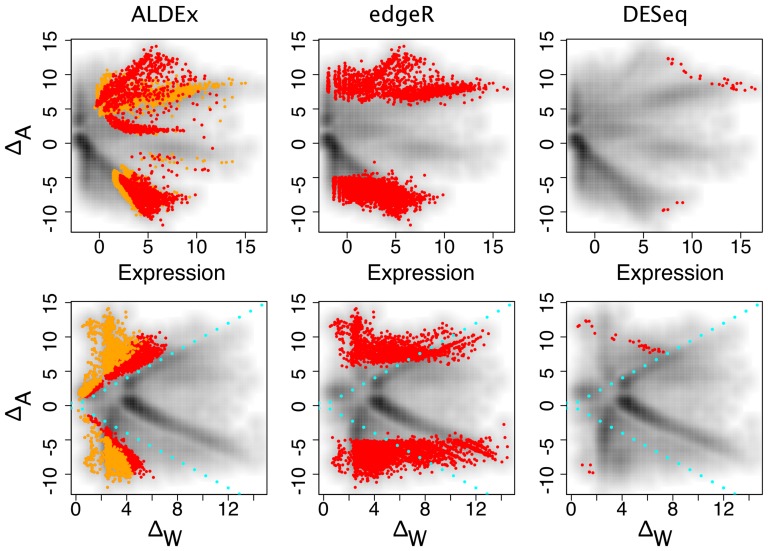
Experimental variance is a major challenge when dealing with high-throughput sequencing data. This variance has several sources: sampling replication, technical replication, variability within biological conditions, and variability between biological conditions. The high per-sample cost of RNA-Seq often precludes the large number of experiments needed to partition observed variance into these categories as per standard ANOVA models. We show that the partitioning of within-condition to between-condition variation cannot reasonably be ignored, whether in single-organism RNA-Seq or in Meta-RNA-Seq experiments, and further find that commonly-used RNA-Seq analysis tools, as described in the literature, do not enforce the constraint that the sum of relative expression levels must be one, and thus report expression levels that are systematically distorted. These two factors lead to misleading inferences if not properly accommodated. As it is usually only the biological between-condition and within-condition differences that are of interest, we developed ALDEx, an ANOVA-like differential expression procedure, to identify genes with greater between- to within-condition differences. We show that the presence of differential expression and the magnitude of these comparative differences can be reasonably estimated with even very small sample sizes.
当处理高通量测序数据时,实验方差是一个主要的挑战。这种方差有几个来源:采样复制、技术复制、生物条件内的可变性以及生物条件之间的可变性。RNA-Seq 的每个样本成本很高,通常无法按照标准的 ANOVA 模型将观察到的方差分成这些类别。我们表明,无论在单个生物体的 RNA-Seq 还是在 Meta-RNA-Seq 实验中,都不能合理地忽略条件内的方差分配到条件之间的变化,进一步发现文献中描述的常用 RNA-Seq 分析工具没有强制实施相对表达水平之和必须为一的约束,因此报告的表达水平会受到系统扭曲。如果不妥善处理这两个因素,就会导致误导性的推断。由于通常只有生物学上的条件之间和条件内的差异是感兴趣的,因此我们开发了 ALDEx,一种类似于 ANOVA 的差异表达程序,用于识别具有更大条件之间与条件内差异的基因。我们表明,即使在非常小的样本量下,也可以合理地估计差异表达的存在及其比较差异的幅度。