Department of Food Science and Technology, University of Nebraska, Lincoln, Nebraska, United States of America.
PLoS One. 2013 Jul 16;8(7):e69621. doi: 10.1371/journal.pone.0069621. Print 2013.
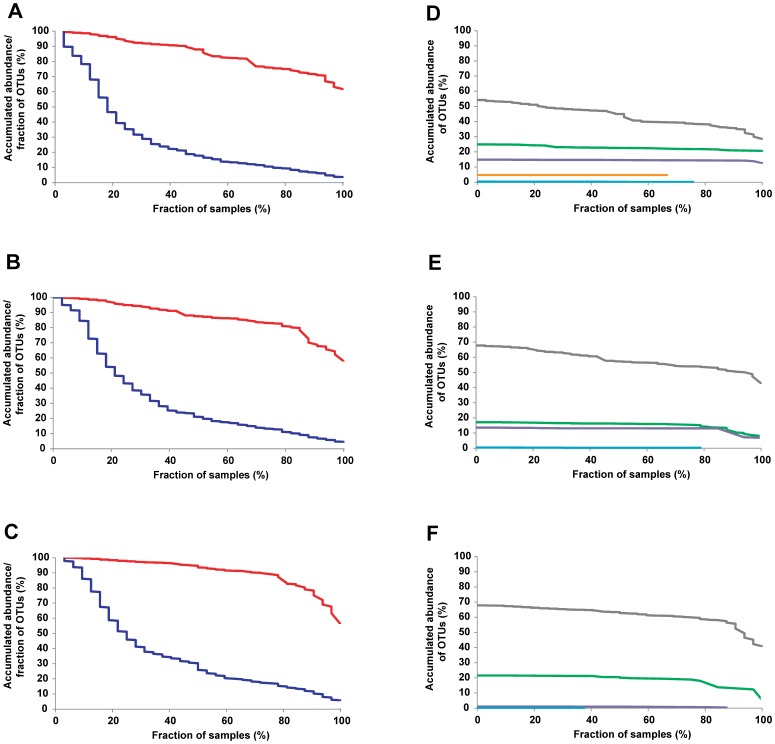
Next-generation sequencing has greatly contributed to an improved ecological understanding of the human gut microbiota. Nevertheless, questions remain regarding the characteristics of this ecosystem and the ecological processes that shape it, and controversy has arisen regarding the stability of the bacterial populations and the existence of a temporal core. In this study, we have characterized the fecal microbial communities of three human individuals over a one-year period by 454 pyrosequencing of 16S rRNA tags in order to investigate the temporal characteristics of the bacterial communities. The findings revealed a temporal core of 33 to 40 species-level Operational Taxonomic Units (OTUs) within subjects. Although these OTUs accounted only for around 12% of the total OTUs detected, they added up to >75% of the total sequences obtained for each individual. In order to determine the capacity of the sequencing and bioinformatic approaches applied during this study to accurately determine the proportion of a core microbiota, we analyzed the fecal microbiota of nine mice with a defined three-member community. This experiment revealed that the sequencing approach inflated the amount of rare OTUs, which introduced a significant degree of artificial variation across samples, and hence reduced the apparent fraction of shared OTUs. However, when assessing the data quantitatively by focusing on dominant lineages, the sequencing approaches deliver an accurate representation of the community. In conclusion, this study revealed that the human fecal microbiota is dominated by around 40 species that maintain persistent populations over the duration of one year. The findings allow conclusions about the ecological factors that shape the community and support the concept of a homeostatic ecosystem controlled largely by deterministic processes. Our analysis of a three-member community revealed that methodological artifacts of OTU-based approaches complicate core calculations, and these limitations have to be considered in the interpretation of microbiome studies.
下一代测序技术极大地促进了人类肠道微生物群落的生态理解。然而,关于这个生态系统的特征以及塑造它的生态过程仍存在问题,并且关于细菌种群的稳定性和存在时间核心的争议也已经出现。在这项研究中,我们通过对 16S rRNA 标签进行 454 焦磷酸测序,对三个人的粪便微生物群落进行了为期一年的特征描述,以研究细菌群落的时间特征。研究结果显示,在个体内部存在 33 到 40 种操作分类单元(OTU)的时间核心。尽管这些 OTU 仅占总检测到的 OTU 的约 12%,但它们加起来占每个人获得的总序列的>75%。为了确定在这项研究中应用的测序和生物信息学方法准确确定核心微生物群比例的能力,我们分析了具有定义的三成员群落的九只小鼠的粪便微生物群。该实验表明,测序方法会增加稀有 OTU 的数量,这会在样本之间引入显著的人为变异,从而降低共享 OTU 的明显比例。然而,当通过关注主导谱系对数据进行定量评估时,测序方法可以准确地表示群落。总之,这项研究表明,人类粪便微生物群主要由大约 40 种物种组成,这些物种在一年的时间内保持着持续的种群。这些发现使我们能够得出关于塑造群落的生态因素的结论,并支持由确定性过程主要控制的稳态生态系统的概念。我们对三成员群落的分析表明,基于 OTU 的方法的方法学人工制品使核心计算变得复杂,在解释微生物组研究时必须考虑这些局限性。