Instituto de Medicina Tropical de São Paulo e Faculdade de Medicina, Departamento de Moléstias Infecciosas e Parasitárias (LIMHC), Universidade de São Paulo, São Paulo, Brazil.
PLoS One. 2013 Aug 2;8(8):e70318. doi: 10.1371/journal.pone.0070318. Print 2013.
High genetic diversity at both inter- and intra-host level are hallmarks of RNA viruses due to the error-prone nature of their genome replication. Several groups have evaluated the extent of viral variability using different RNA virus deep sequencing methods. Although much of this effort has been dedicated to pathogens that cause chronic infections in humans, few studies investigated arthropod-borne, acute viral infections.
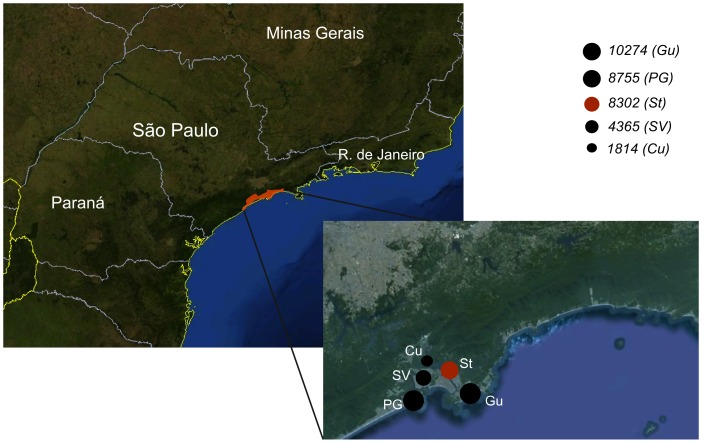
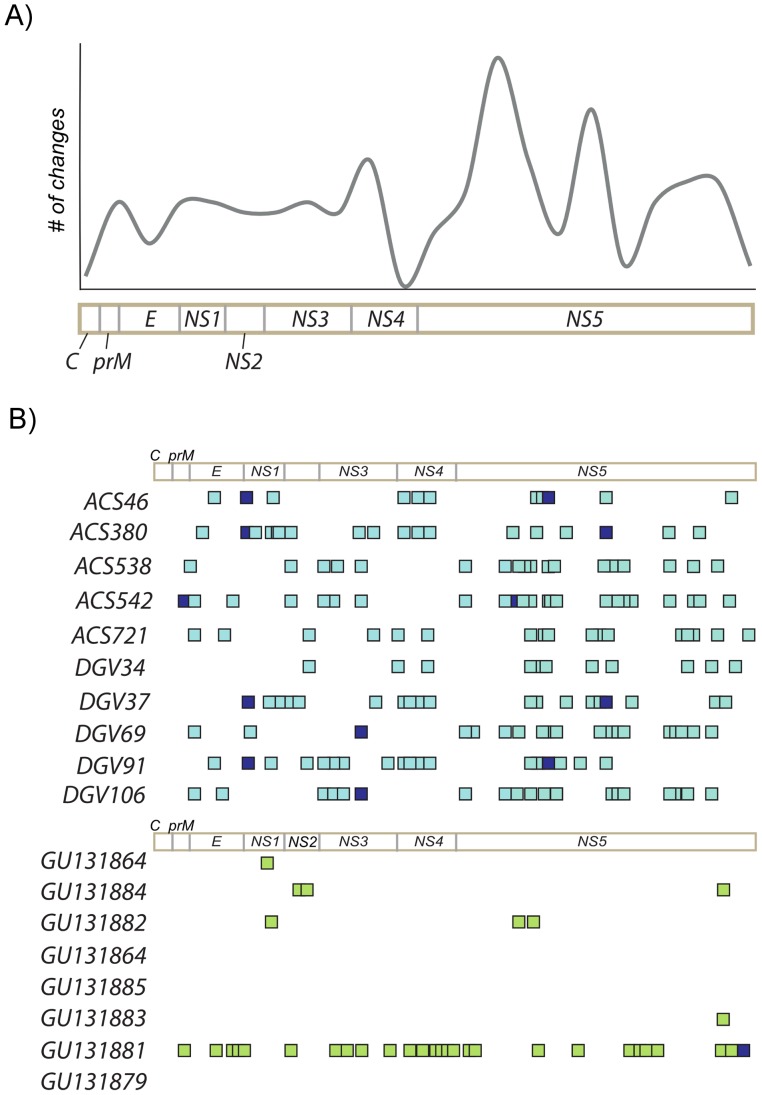
We deep sequenced the complete genome of ten DENV2 isolates from representative classical and severe cases sampled in a large outbreak in Brazil using two different approaches. Analysis of the consensus genomes confirmed the larger extent of the 2010 epidemic in comparison to a previous epidemic caused by the same viruses in another city two years before (genetic distance = 0.002 and 0.0008 respectively). Analysis of viral populations within the host revealed a high level of conservation. After excluding homopolymer regions of 454/Roche generated sequences, we found 10 to 44 variable sites per genome population at a frequency of >1%, resulting in very low intra-host genetic diversity. While up to 60% of all variable sites at intra-host level were non-synonymous changes, only 10% of inter-host variability resulted from non-synonymous mutations, indicative of purifying selection at the population level.
Despite the error-prone nature of RNA-dependent RNA-polymerase, dengue viruses maintain low levels of intra-host variability.
由于其基因组复制的易错性质,RNA 病毒在宿主间和宿主内均具有高度的遗传多样性。有几个小组使用不同的 RNA 病毒深度测序方法评估了病毒变异性的程度。尽管其中大部分工作都致力于研究导致人类慢性感染的病原体,但很少有研究调查过节肢动物传播的急性病毒感染。
我们使用两种不同的方法对来自巴西大规模暴发的具有代表性的典型和严重病例的 10 个 DENV2 分离株的完整基因组进行了深度测序。对共识基因组的分析证实,与两年前同一病毒在另一个城市引起的上一次流行相比,2010 年的流行程度更大(遗传距离分别为 0.002 和 0.0008)。对宿主内病毒群体的分析显示出高度的保守性。在排除 454/Roche 生成序列的同源多聚体区域后,我们发现每个基因组群体中有 10 到 44 个频率大于 1%的可变位点,导致宿主内遗传多样性非常低。虽然在宿主内水平上高达 60%的所有可变位点是非同义变化,但只有 10%的宿主间变异性是由非同义突变引起的,表明在群体水平上存在纯化选择。
尽管 RNA 依赖性 RNA 聚合酶具有易错性质,但登革热病毒仍保持低水平的宿主内变异性。