Michele Di Pierro, Ron Elber
Institute for Computational Engineering and Sciences, University of Texas at Austin, Austin TX 78712.
J Chem Theory Comput. 2013 Aug 13;9(8):3311-3320. doi: 10.1021/ct400313n.
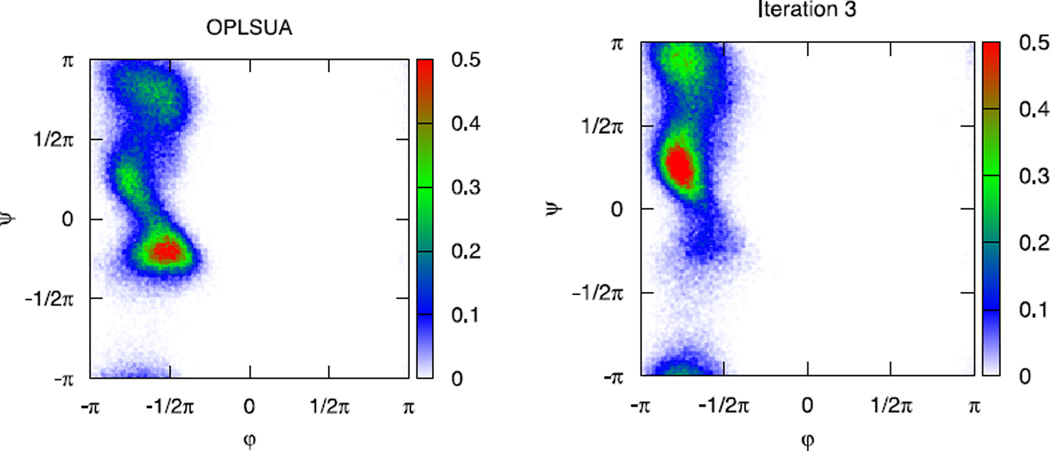
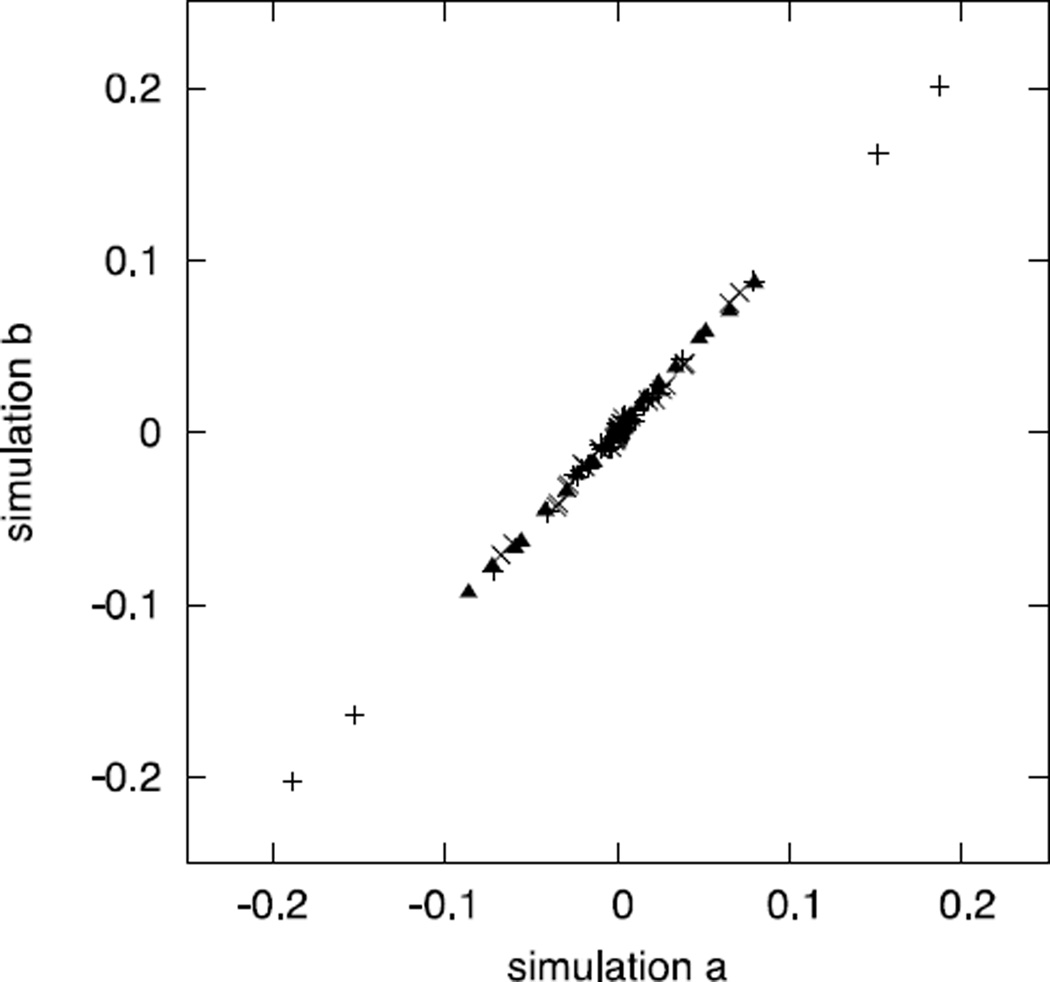
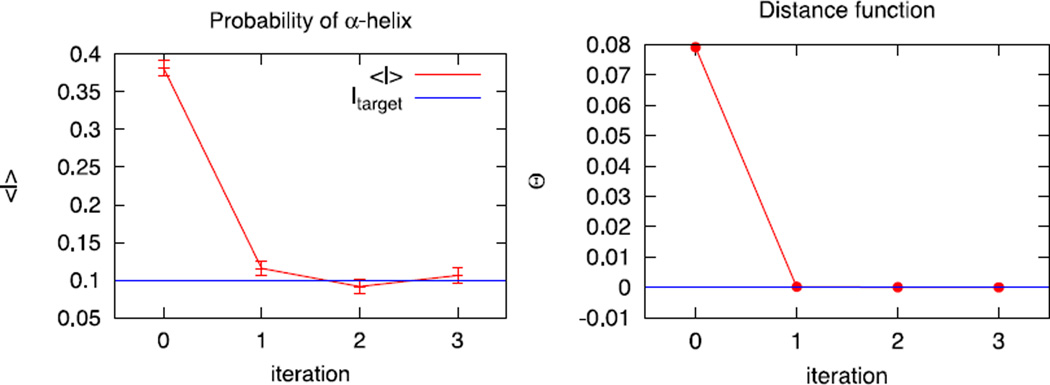
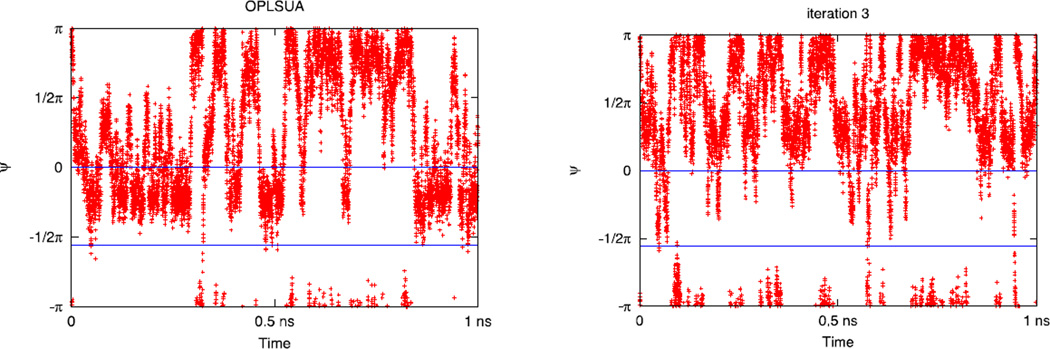
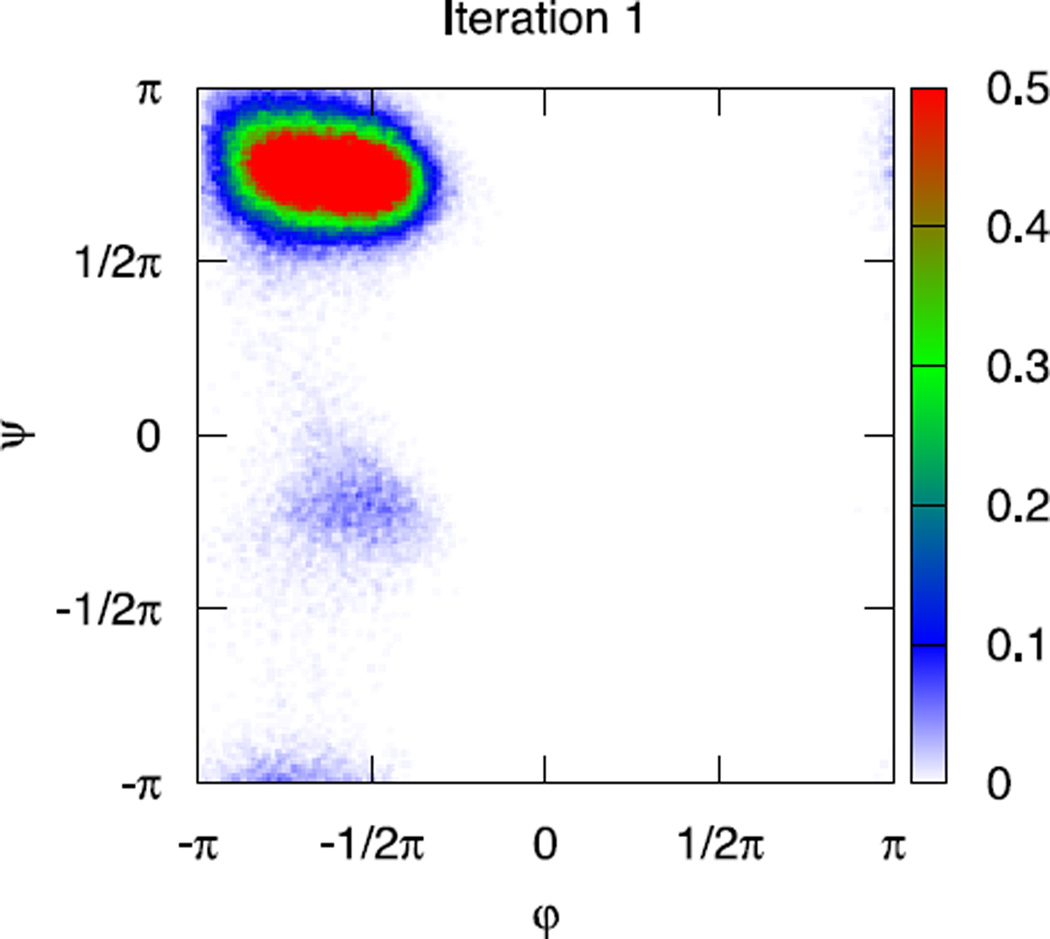
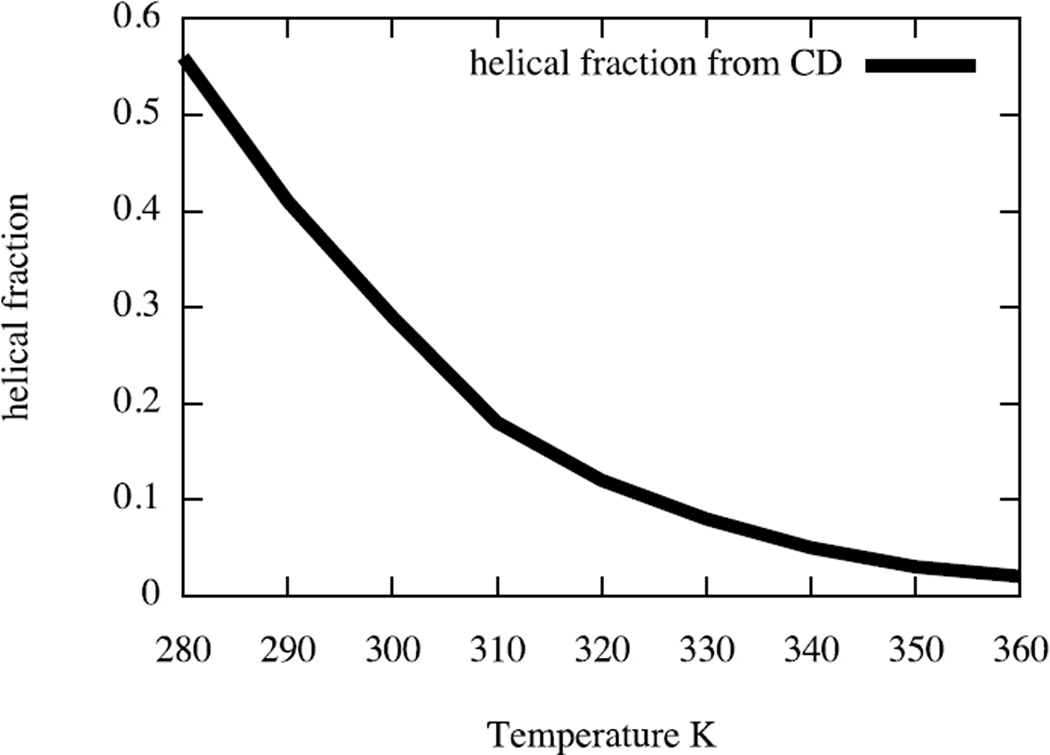
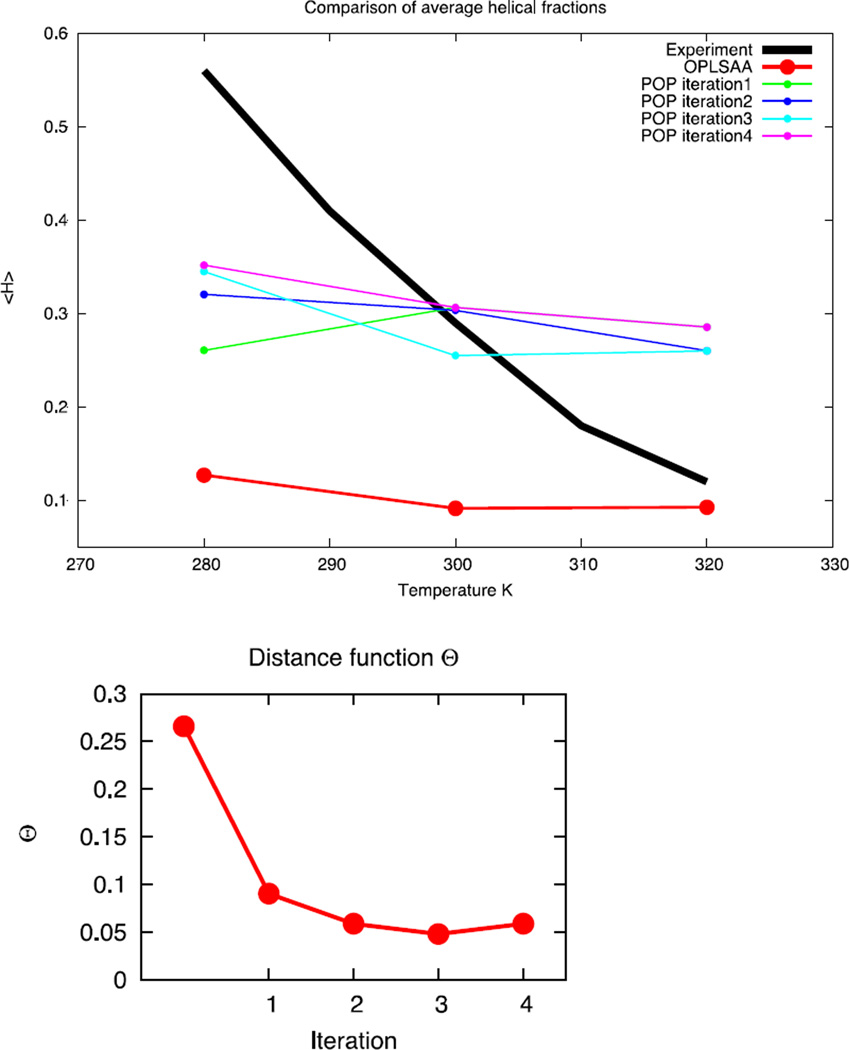
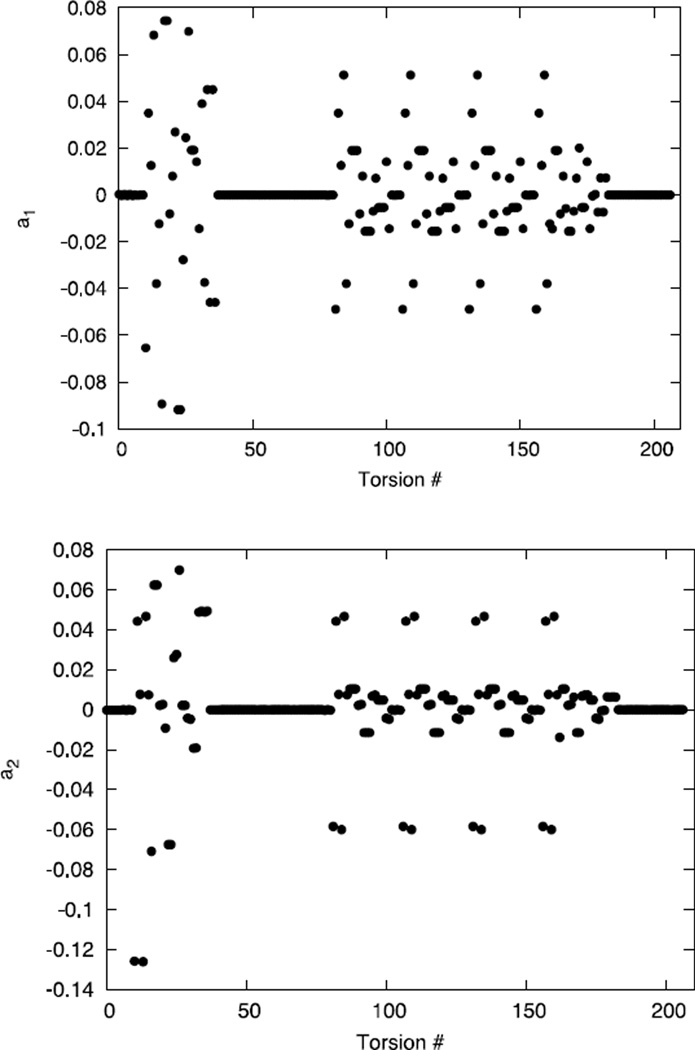
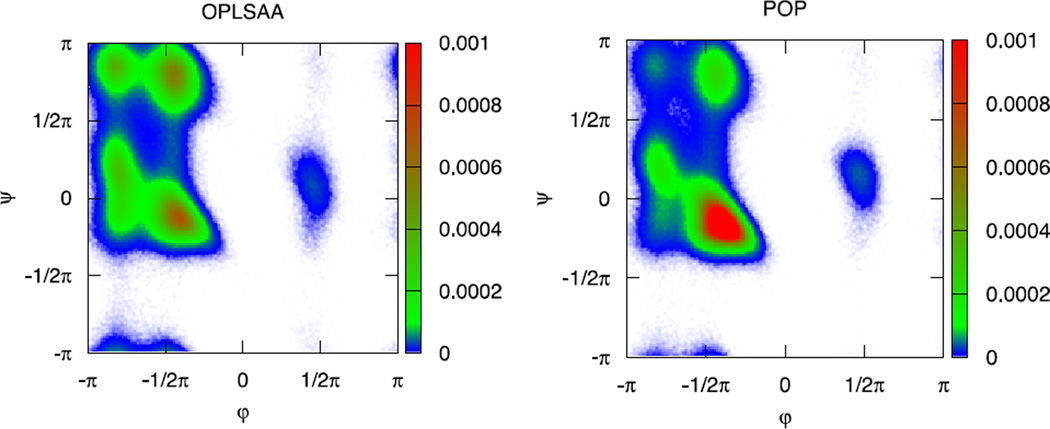
An algorithm and software to refine parameters of empirical energy functions according to condensed phase experimental measurements are discussed. The algorithm is based on sensitivity analysis and local minimization of the differences between experiment and simulation as a function of potential parameters. It is illustrated for a toy problem of alanine dipeptide and is applied to folding of the peptide WAAAH. The helix fraction is highly sensitive to the potential parameters while the slope of the melting curve is not. The sensitivity variations make it difficult to satisfy both observations simultaneously. We conjecture that there is no set of parameters that reproduces experimental melting curves of short peptides that are modeled with the usual functional form of a force field.
本文讨论了一种根据凝聚相实验测量结果来优化经验能量函数参数的算法及软件。该算法基于灵敏度分析以及将实验与模拟之间的差异作为势能参数的函数进行局部最小化。通过丙氨酸二肽的一个简单问题进行了说明,并应用于肽WAAAH的折叠。螺旋分数对势能参数高度敏感,而熔解曲线的斜率则不然。灵敏度的变化使得难以同时满足这两个观测结果。我们推测,不存在一组参数能够重现用通常力场函数形式建模的短肽的实验熔解曲线。