Wellcome Trust Sanger Institute, Hinxton, UK.
Wellcome Trust Sanger Institute, Hinxton, UK; Division of Infection and Immunity, University College London, London, UK.
Lancet. 2013 Dec 14;382(9909):1993-2002. doi: 10.1016/S0140-6736(13)61887-5. Epub 2013 Sep 20.
Since June, 2012, Middle East respiratory syndrome coronavirus (MERS-CoV) has, worldwide, caused 104 infections in people including 49 deaths, with 82 cases and 41 deaths reported from Saudi Arabia. In addition to confirming diagnosis, we generated the MERS-CoV genomic sequences obtained directly from patient samples to provide important information on MERS-CoV transmission, evolution, and origin.
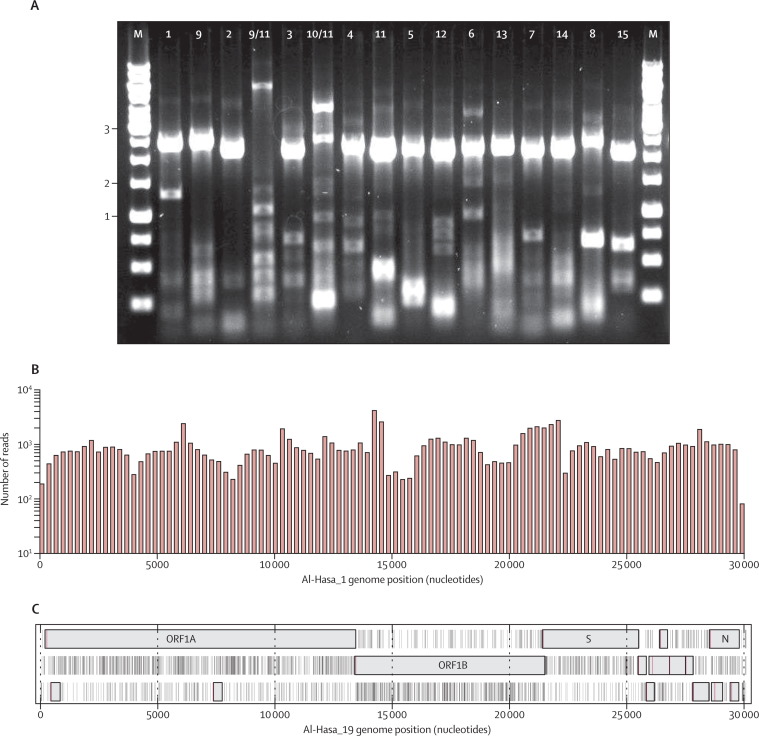
Full genome deep sequencing was done on nucleic acid extracted directly from PCR-confirmed clinical samples. Viral genomes were obtained from 21 MERS cases of which 13 had 100%, four 85-95%, and four 30-50% genome coverage. Phylogenetic analysis of the 21 sequences, combined with nine published MERS-CoV genomes, was done.
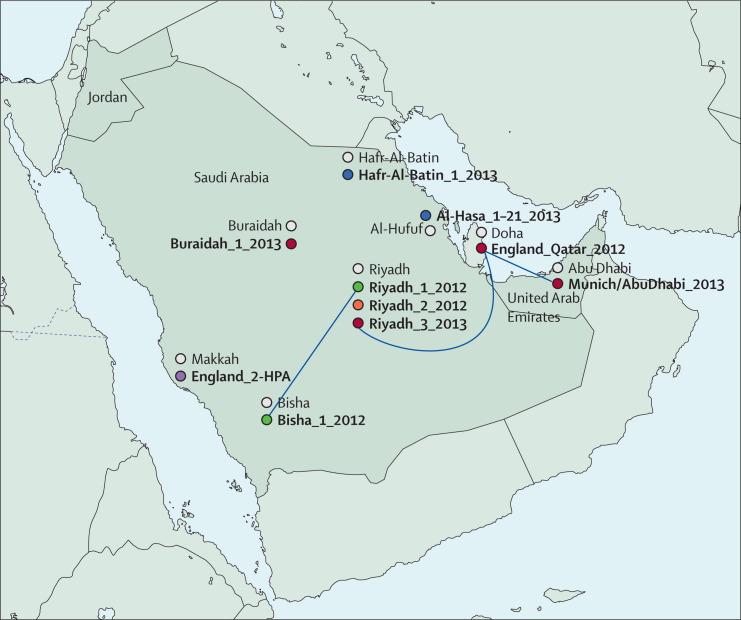
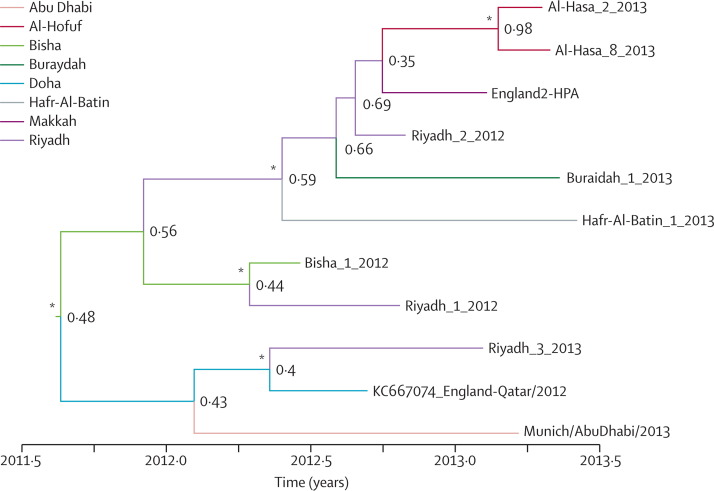
Three distinct MERS-CoV genotypes were identified in Riyadh. Phylogeographic analyses suggest the MERS-CoV zoonotic reservoir is geographically disperse. Selection analysis of the MERS-CoV genomes reveals the expected accumulation of genetic diversity including changes in the S protein. The genetic diversity in the Al-Hasa cluster suggests that the hospital outbreak might have had more than one virus introduction.
We present the largest number of MERS-CoV genomes (21) described so far. MERS-CoV full genome sequences provide greater detail in tracking transmission. Multiple introductions of MERS-CoV are identified and suggest lower R0 values. Transmission within Saudi Arabia is consistent with either movement of an animal reservoir, animal products, or movement of infected people. Further definition of the exposures responsible for the sporadic introductions of MERS-CoV into human populations is urgently needed.
Saudi Arabian Ministry of Health, Wellcome Trust, European Community, and National Institute of Health Research University College London Hospitals Biomedical Research Centre.
自 2012 年 6 月以来,中东呼吸综合征冠状病毒(MERS-CoV)已在全球范围内导致 104 人感染,其中包括 49 人死亡,其中 82 例和 41 例死亡报告来自沙特阿拉伯。除了确诊诊断外,我们还直接从患者样本中生成了 MERS-CoV 基因组序列,为 MERS-CoV 的传播、进化和起源提供了重要信息。
对直接从 PCR 确认的临床样本中提取的核酸进行全基因组深度测序。从 21 例 MERS 病例中获得了病毒基因组,其中 13 例覆盖率为 100%,4 例覆盖率为 85-95%,4 例覆盖率为 30-50%。对 21 个序列进行了系统发育分析,并结合 9 个已发表的 MERS-CoV 基因组进行了分析。
在利雅得确定了三种不同的 MERS-CoV 基因型。系统地理学分析表明,MERS-CoV 的动物宿主是地理上分散的。对 MERS-CoV 基因组的选择分析表明,遗传多样性包括 S 蛋白的变化在内,预计会有所积累。Al-Hasa 群集中的遗传多样性表明,医院爆发可能不止有一次病毒传入。
我们目前描述了数量最多的 MERS-CoV 基因组(21 个)。MERS-CoV 全基因组序列提供了更详细的跟踪传播情况。确定了多次 MERS-CoV 传入,表明 R0 值较低。沙特阿拉伯境内的传播情况与动物宿主的移动、动物产品的移动或受感染人群的移动一致。迫切需要进一步确定导致 MERS-CoV 偶尔传入人群的暴露源。
沙特阿拉伯卫生部、惠康信托基金会、欧洲共同体和英国国民健康保险制度大学学院伦敦医院生物医学研究中心。