Cotten Matthew, Watson Simon J, Zumla Alimuddin I, Makhdoom Hatem Q, Palser Anne L, Ong Swee Hoe, Al Rabeeah Abdullah A, Alhakeem Rafat F, Assiri Abdullah, Al-Tawfiq Jaffar A, Albarrak Ali, Barry Mazin, Shibl Atef, Alrabiah Fahad A, Hajjar Sami, Balkhy Hanan H, Flemban Hesham, Rambaut Andrew, Kellam Paul, Memish Ziad A
mBio. 2014 Feb 18;5(1):e01062-13. doi: 10.1128/mBio.01062-13.
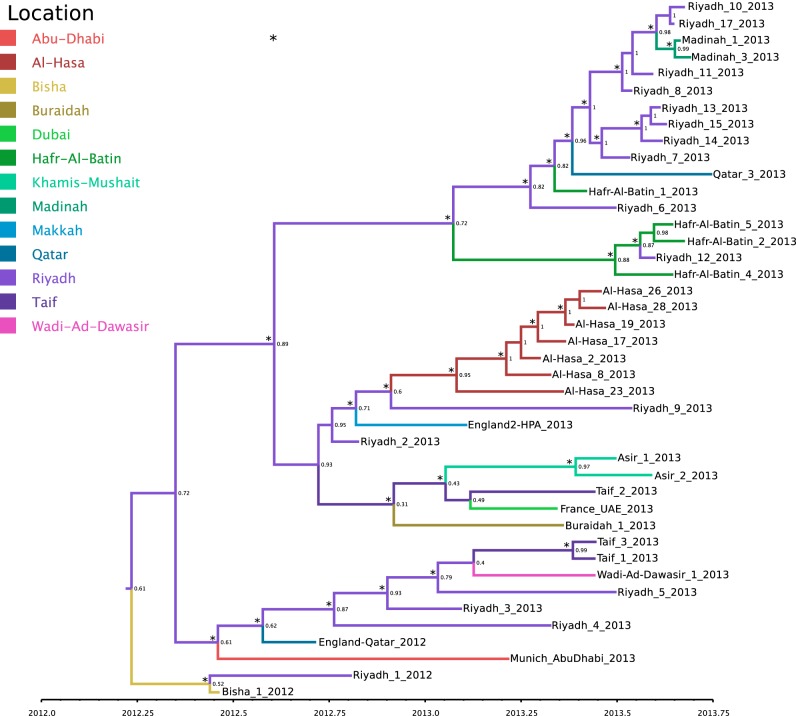
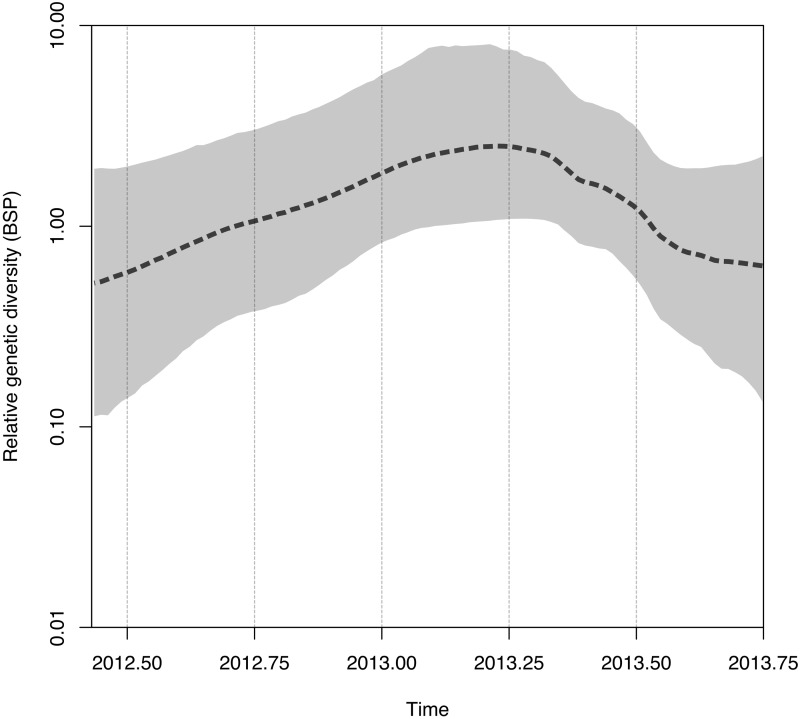
The Middle East respiratory syndrome coronavirus (MERS-CoV) was first documented in the Kingdom of Saudi Arabia (KSA) in 2012 and, to date, has been identified in 180 cases with 43% mortality. In this study, we have determined the MERS-CoV evolutionary rate, documented genetic variants of the virus and their distribution throughout the Arabian peninsula, and identified the genome positions under positive selection, important features for monitoring adaptation of MERS-CoV to human transmission and for identifying the source of infections. Respiratory samples from confirmed KSA MERS cases from May to September 2013 were subjected to whole-genome deep sequencing, and 32 complete or partial sequences (20 were ≥ 99% complete, 7 were 50 to 94% complete, and 5 were 27 to 50% complete) were obtained, bringing the total available MERS-CoV genomic sequences to 65. An evolutionary rate of 1.12 × 10(-3) substitutions per site per year (95% credible interval [95% CI], 8.76 × 10(-4); 1.37 × 10(-3)) was estimated, bringing the time to most recent common ancestor to March 2012 (95% CI, December 2011; June 2012). Only one MERS-CoV codon, spike 1020, located in a domain required for cell entry, is under strong positive selection. Four KSA MERS-CoV phylogenetic clades were found, with 3 clades apparently no longer contributing to current cases. The size of the population infected with MERS-CoV showed a gradual increase to June 2013, followed by a decline, possibly due to increased surveillance and infection control measures combined with a basic reproduction number (R0) for the virus that is less than 1.
MERS-CoV adaptation toward higher rates of sustained human-to-human transmission appears not to have occurred yet. While MERS-CoV transmission currently appears weak, careful monitoring of changes in MERS-CoV genomes and of the MERS epidemic should be maintained. The observation of phylogenetically related MERS-CoV in geographically diverse locations must be taken into account in efforts to identify the animal source and transmission of the virus.
中东呼吸综合征冠状病毒(MERS-CoV)于2012年首次在沙特阿拉伯王国(KSA)被记录,迄今为止,已确诊180例,死亡率达43%。在本研究中,我们确定了MERS-CoV的进化速率,记录了该病毒的基因变异及其在阿拉伯半岛的分布情况,并确定了受到正选择的基因组位置,这些都是监测MERS-CoV对人际传播的适应性以及确定感染源的重要特征。对2013年5月至9月确诊的沙特阿拉伯MERS病例的呼吸道样本进行全基因组深度测序,获得了32个完整或部分序列(20个序列完整性≥99%,7个序列完整性为50%至94%,5个序列完整性为27%至50%),使可用的MERS-CoV基因组序列总数达到65个。估计进化速率为每年每个位点1.12×10⁻³个替换(95%可信区间[95%CI],8.76×10⁻⁴;1.37×10⁻³),最近共同祖先的时间为2012年3月(95%CI,2011年12月;2012年6月)。只有一个MERS-CoV密码子,即位于细胞进入所需结构域的刺突蛋白1020,受到强烈正选择。发现了四个沙特阿拉伯MERS-CoV系统发育分支,其中3个分支显然不再导致当前病例。感染MERS-CoV的人群规模在2013年6月前逐渐增加,随后下降,这可能是由于监测和感染控制措施加强,以及该病毒的基本再生数(R₀)小于1。
MERS-CoV似乎尚未适应更高的持续人际传播率。虽然目前MERS-CoV的传播似乎较弱,但仍应密切监测MERS-CoV基因组的变化和MERS疫情。在确定病毒的动物源和传播途径时,必须考虑在不同地理位置观察到的系统发育相关的MERS-CoV。