1] Graduate Program in Applied Mathematics & Statistics, and Scientific Computation, University of Maryland, College Park, Maryland, USA. [2] Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland, USA.
Nat Methods. 2013 Dec;10(12):1200-2. doi: 10.1038/nmeth.2658. Epub 2013 Sep 29.
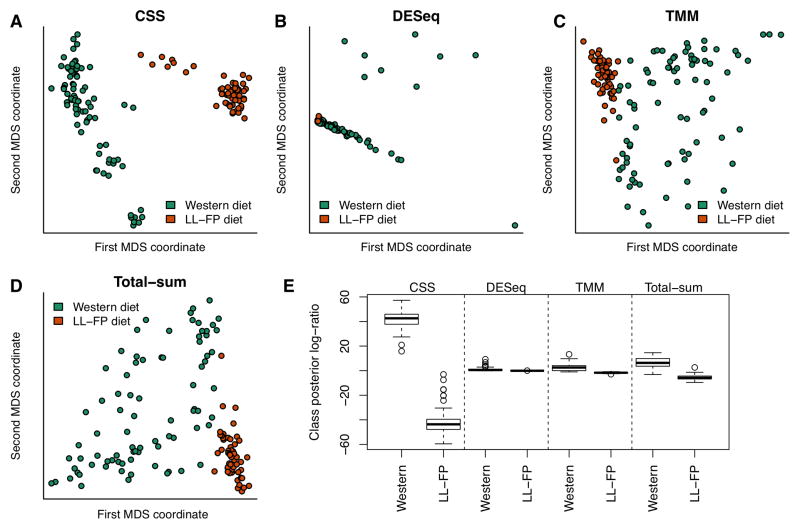
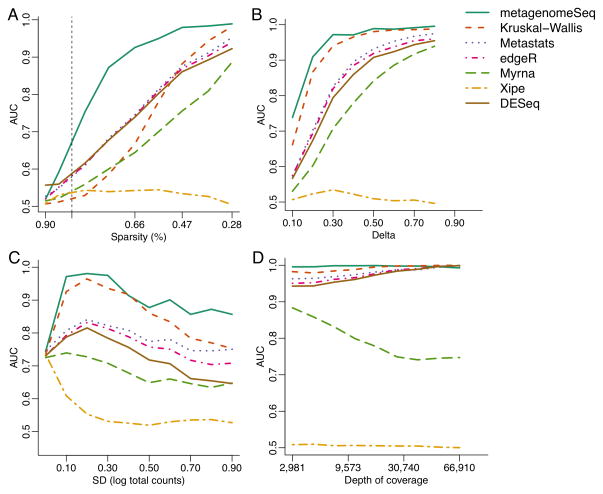
We introduce a methodology to assess differential abundance in sparse high-throughput microbial marker-gene survey data. Our approach, implemented in the metagenomeSeq Bioconductor package, relies on a novel normalization technique and a statistical model that accounts for undersampling-a common feature of large-scale marker-gene studies. Using simulated data and several published microbiota data sets, we show that metagenomeSeq outperforms the tools currently used in this field.
我们介绍了一种评估稀疏高通量微生物标记基因调查数据中丰度差异的方法。我们的方法,在 metagenomeSeq Bioconductor 包中实现,依赖于一种新颖的归一化技术和一个统计模型,该模型考虑了欠采样——这是大规模标记基因研究的一个常见特征。使用模拟数据和几个已发表的微生物组数据集,我们表明 metagenomeSeq 优于该领域目前使用的工具。