Lauss Martin, Visne Ilhami, Kriegner Albert, Ringnér Markus, Jönsson Göran, Höglund Mattias
Department of Oncology, Clinical Sciences, Lund University, Sweden.
Cancer Inform. 2013 Sep 23;12:193-201. doi: 10.4137/CIN.S12862. eCollection 2013.
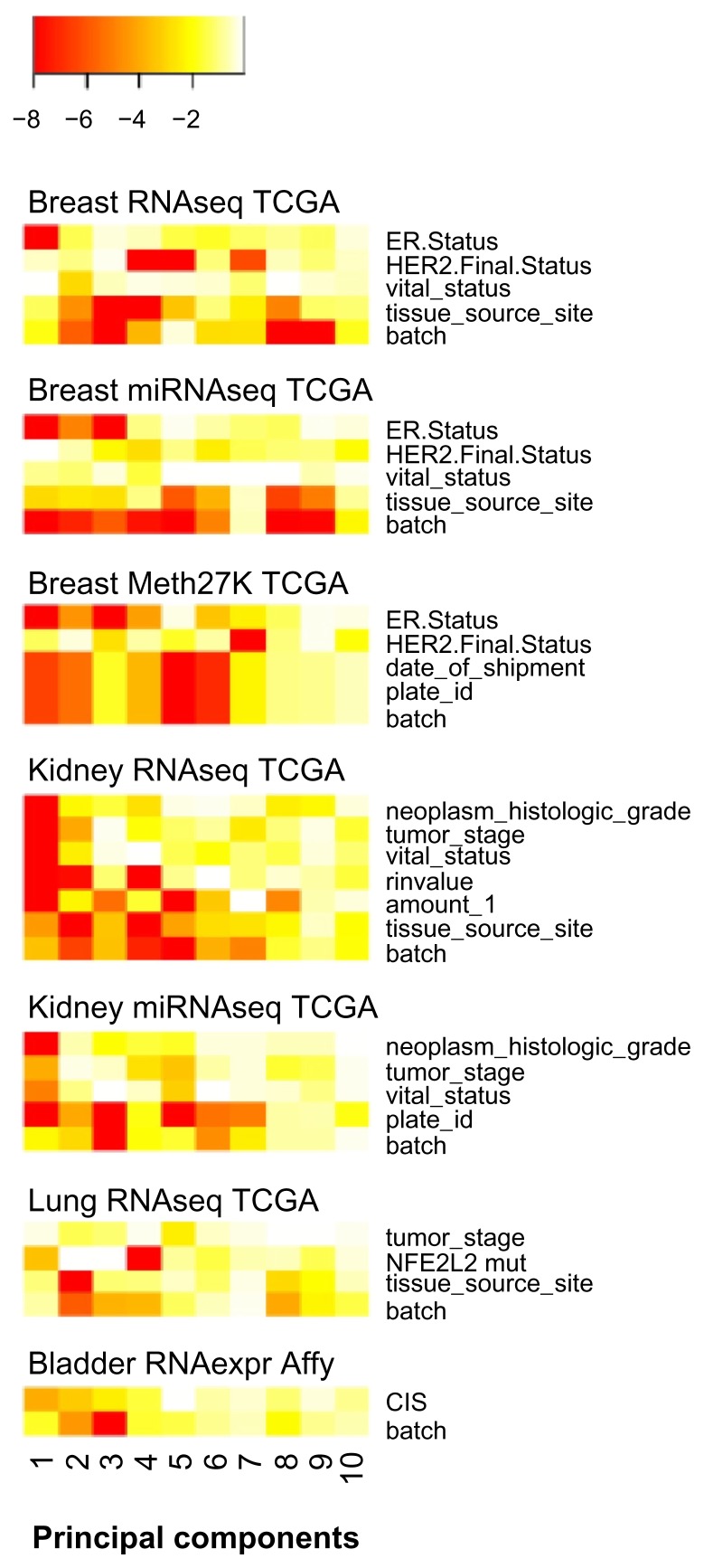
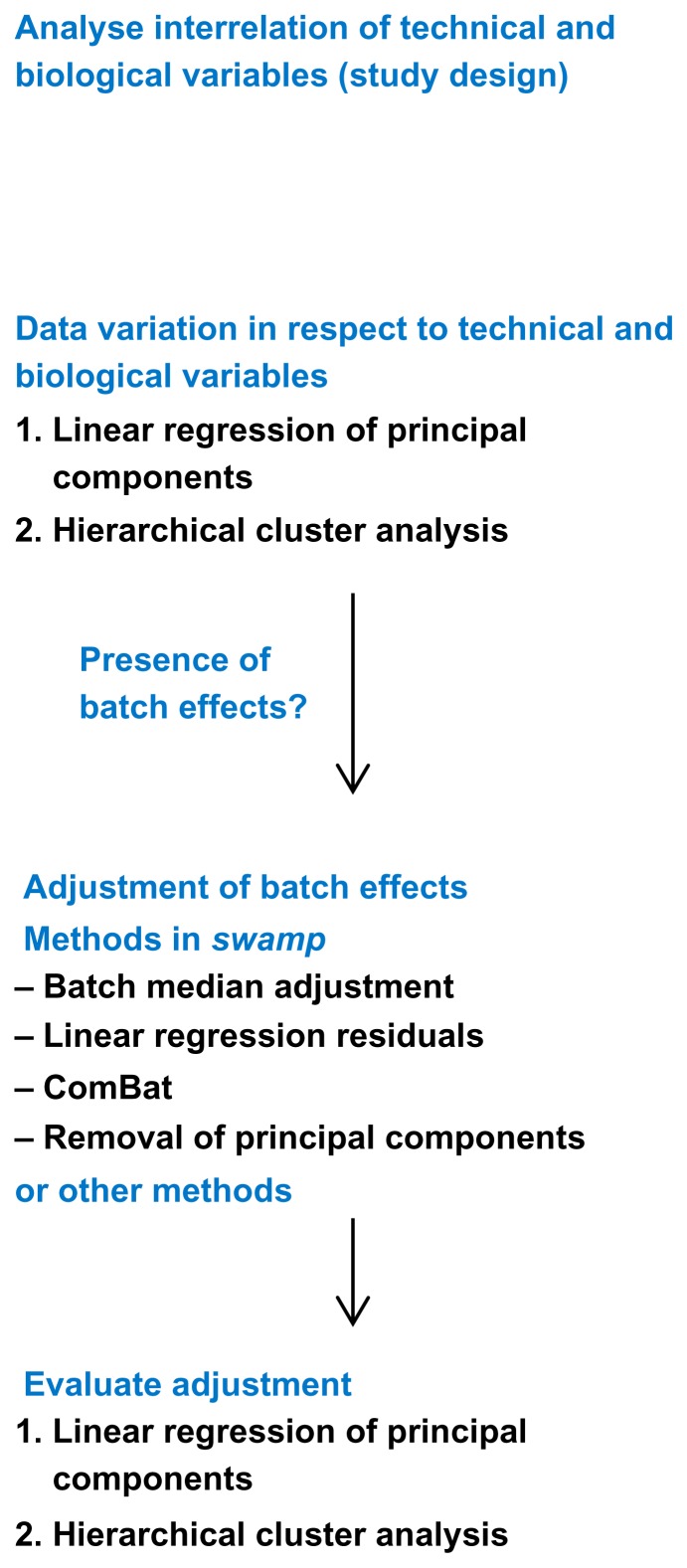
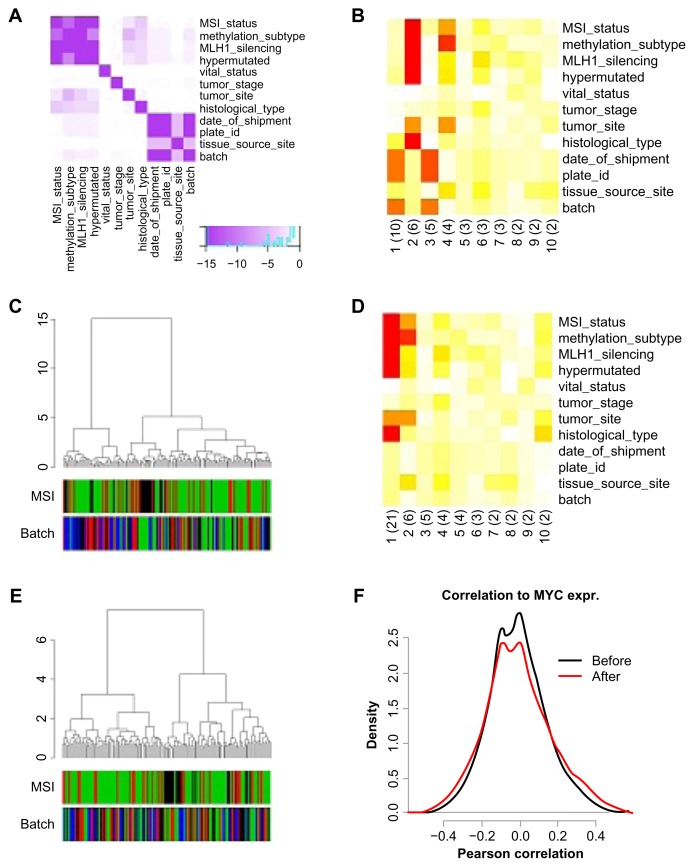
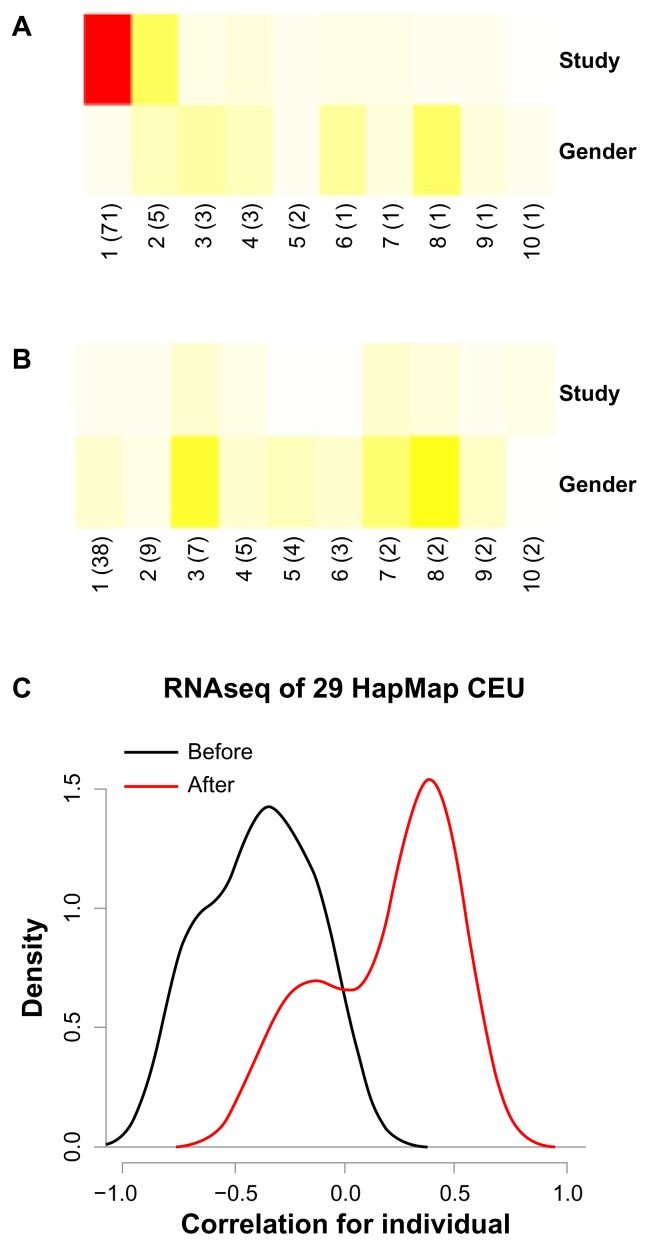
High-dimensional datasets can be confounded by variation from technical sources, such as batches. Undetected batch effects can have severe consequences for the validity of a study's conclusion(s). We evaluate high-throughput RNAseq and miRNAseq as well as DNA methylation and gene expression microarray datasets, mainly from the Cancer Genome Atlas (TCGA) project, in respect to technical and biological annotations. We observe technical bias in these datasets and discuss corrective interventions. We then suggest a general procedure to control study design, detect technical bias using linear regression of principal components, correct for batch effects, and re-evaluate principal components. This procedure is implemented in the R package swamp, and as graphical user interface software. In conclusion, high-throughput platforms that generate continuous measurements are sensitive to various forms of technical bias. For such data, monitoring of technical variation is an important analysis step.
高维数据集可能会因技术来源(如批次)的变异而产生混淆。未检测到的批次效应可能会对研究结论的有效性产生严重影响。我们评估了高通量RNA测序和miRNA测序以及DNA甲基化和基因表达微阵列数据集,主要来自癌症基因组图谱(TCGA)项目,涉及技术和生物学注释。我们在这些数据集中观察到技术偏差并讨论了纠正措施。然后,我们提出了一个通用程序,用于控制研究设计,使用主成分线性回归检测技术偏差,校正批次效应,并重新评估主成分。此程序在R包swamp中实现,并作为图形用户界面软件。总之,生成连续测量值的高通量平台对各种形式的技术偏差很敏感。对于此类数据,监测技术变异是一个重要的分析步骤。