Das Jishnu, Fragoza Robert, Lee Hao Ran, Cordero Nicolas A, Guo Yu, Meyer Michael J, Vo Tommy V, Wang Xiujuan, Yu Haiyuan
Department of Biological Statistics and Computational Biology, Cornell University, Ithaca, NY 14853, USA.
Weill Institute for Cell and Molecular Biology, Cornell University, Ithaca, NY 14853, USA.
Mol Biosyst. 2014 Jan;10(1):9-17. doi: 10.1039/c3mb70225a.
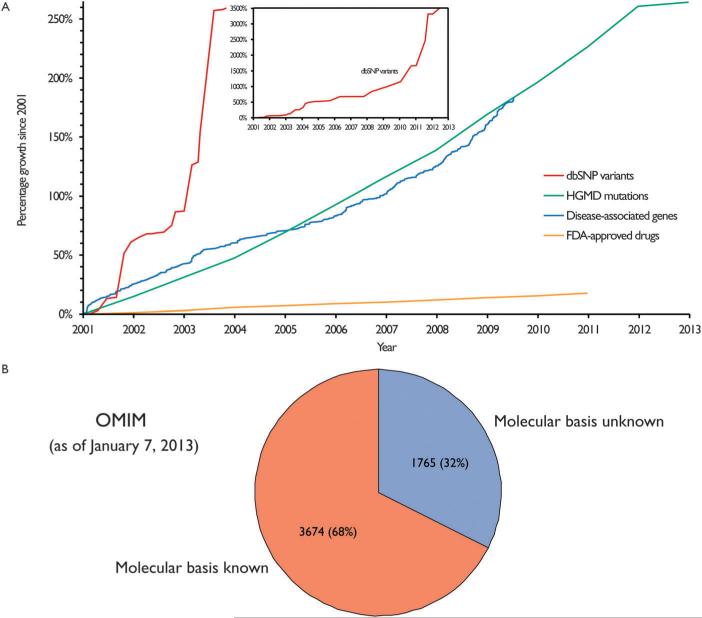
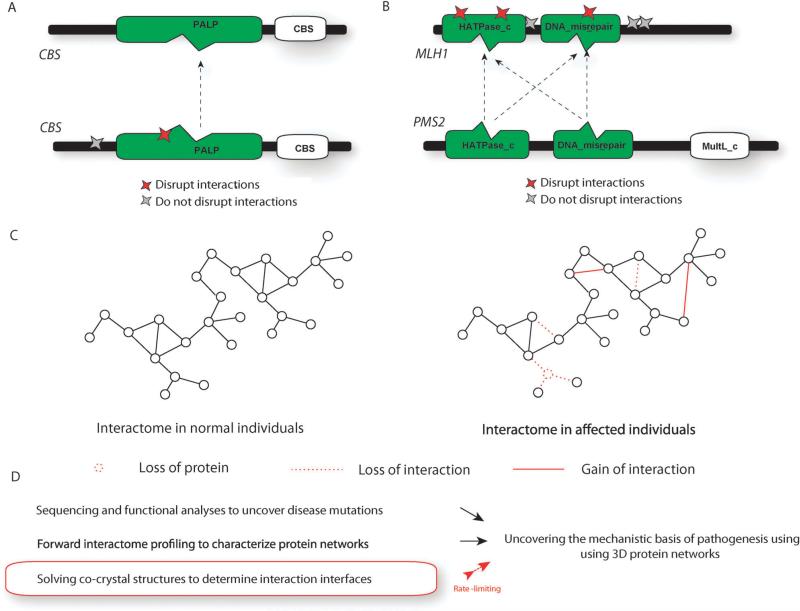
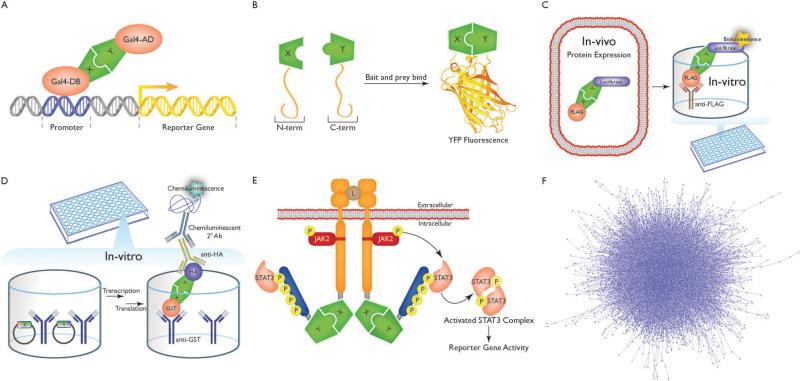
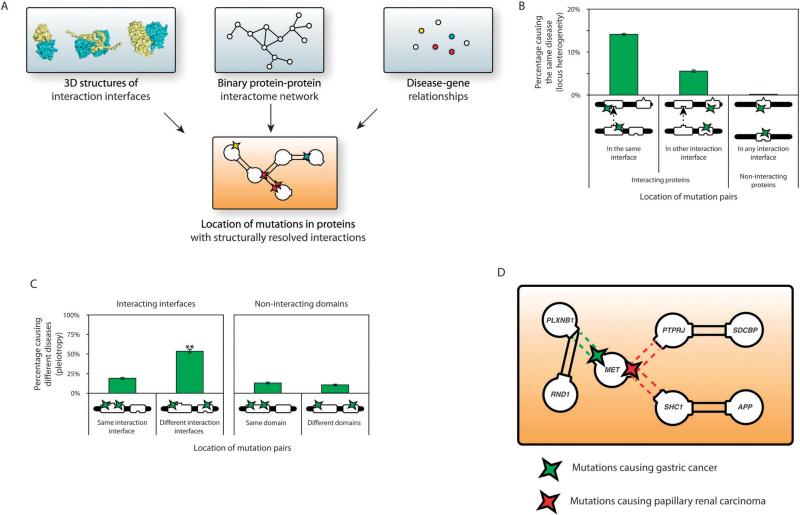
The study of the molecular basis of human disease has gained increasing attention over the past decade. With significant improvements in sequencing efficiency and throughput, a wealth of genotypic data has become available. However the translation of this information into concrete advances in diagnostic and clinical setups has proved far more challenging. Two major reasons for this are the lack of functional annotation for genomic variants and the complex nature of genotype-to-phenotype relationships. One fundamental approach to bypass these issues is to examine the effects of genetic variation at the level of proteins as they are directly involved in carrying out biological functions. Within the cell, proteins function by interacting with other proteins as a part of an underlying interactome network. This network can be determined using interactome mapping - a combination of high-throughput experimental toolkits and curation from small-scale studies. Integrating structural information from co-crystals with the network allows generation of a structurally resolved network. Within the context of this network, the structural principles of disease mutations can be examined and used to generate reliable mechanistic hypotheses regarding disease pathogenesis.
在过去十年中,对人类疾病分子基础的研究越来越受到关注。随着测序效率和通量的显著提高,大量的基因型数据已可获取。然而,将这些信息转化为诊断和临床实践中的具体进展已证明具有更大的挑战性。造成这种情况的两个主要原因是基因组变异缺乏功能注释以及基因型与表型关系的复杂性。绕过这些问题的一种基本方法是在蛋白质水平上研究遗传变异的影响,因为蛋白质直接参与执行生物学功能。在细胞内,蛋白质作为潜在相互作用组网络的一部分,通过与其他蛋白质相互作用发挥功能。这个网络可以使用相互作用组图谱来确定——这是高通量实验工具包和小规模研究的策展的结合。将来自共晶体的结构信息与网络整合,可以生成一个结构解析网络。在这个网络的背景下,可以研究疾病突变的结构原理,并用于生成关于疾病发病机制的可靠机制假设。