Institute for Genome Sciences, University of Maryland School of Medicine, Baltimore, Maryland 21201, USA.
BMC Genomics. 2013 Oct 10;14:693. doi: 10.1186/1471-2164-14-693.
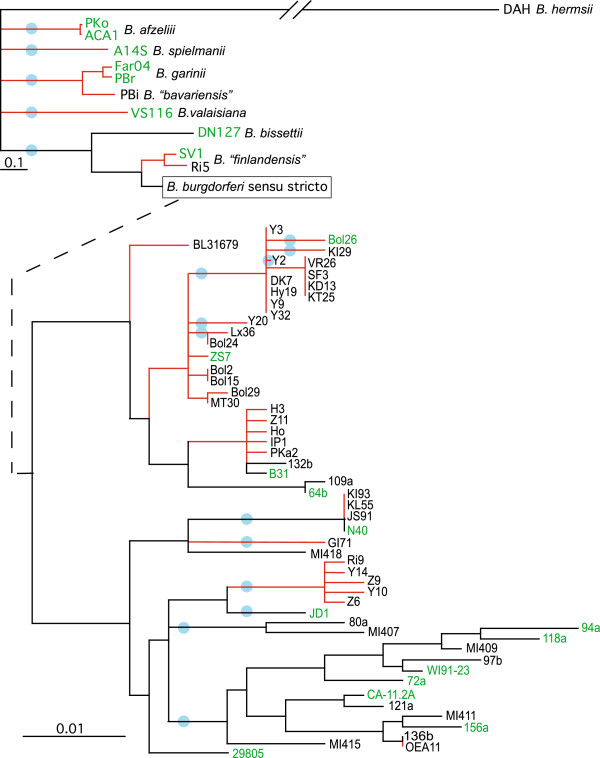
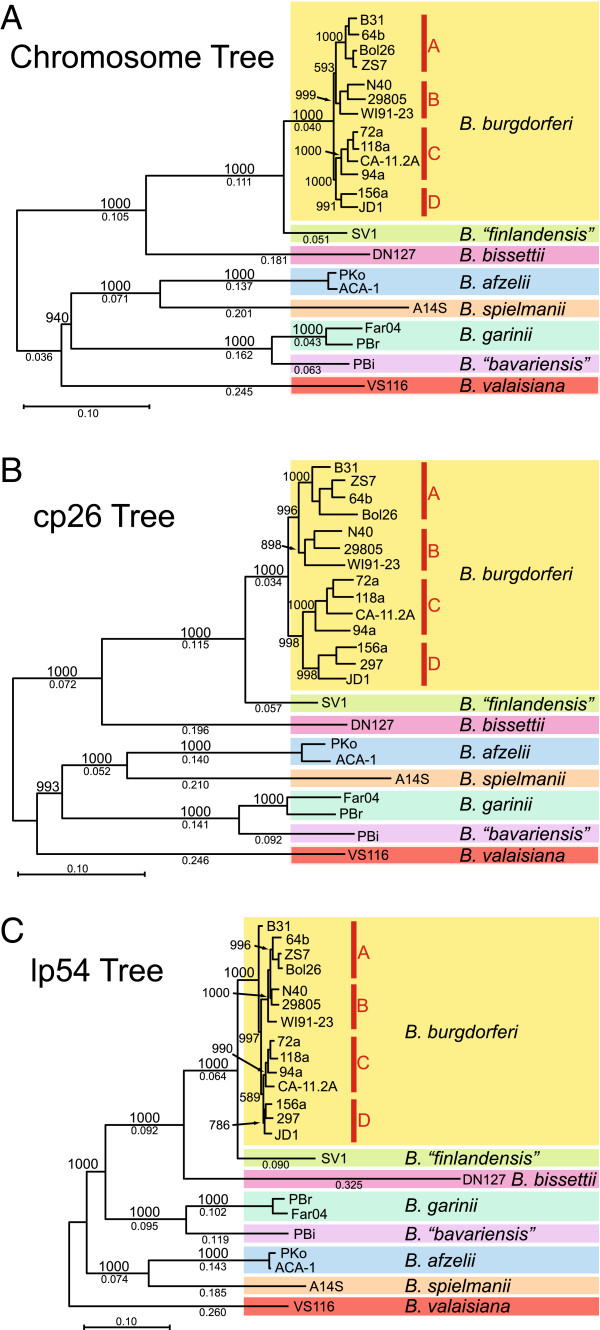
Lyme disease is caused by spirochete bacteria from the Borrelia burgdorferi sensu lato (B. burgdorferi s.l.) species complex. To reconstruct the evolution of B. burgdorferi s.l. and identify the genomic basis of its human virulence, we compared the genomes of 23 B. burgdorferi s.l. isolates from Europe and the United States, including B. burgdorferi sensu stricto (B. burgdorferi s.s., 14 isolates), B. afzelii (2), B. garinii (2), B. "bavariensis" (1), B. spielmanii (1), B. valaisiana (1), B. bissettii (1), and B. "finlandensis" (1).
Robust B. burgdorferi s.s. and B. burgdorferi s.l. phylogenies were obtained using genome-wide single-nucleotide polymorphisms, despite recombination. Phylogeny-based pan-genome analysis showed that the rate of gene acquisition was higher between species than within species, suggesting adaptive speciation. Strong positive natural selection drives the sequence evolution of lipoproteins, including chromosomally-encoded genes 0102 and 0404, cp26-encoded ospC and b08, and lp54-encoded dbpA, a07, a22, a33, a53, a65. Computer simulations predicted rapid adaptive radiation of genomic groups as population size increases.
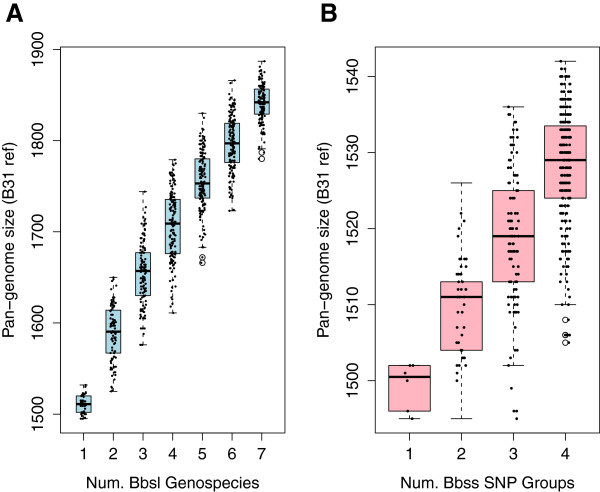
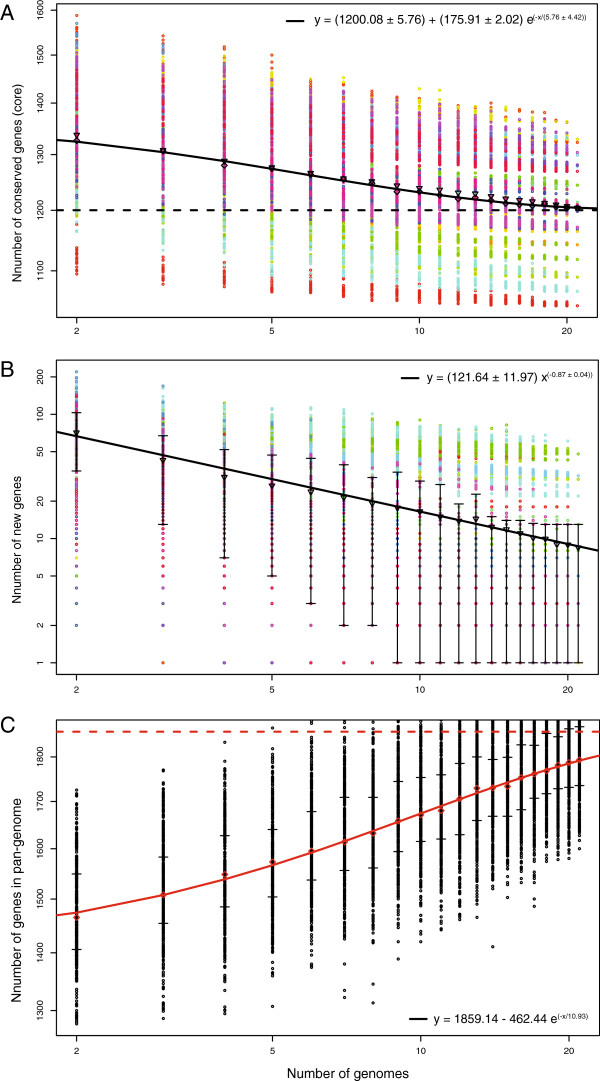
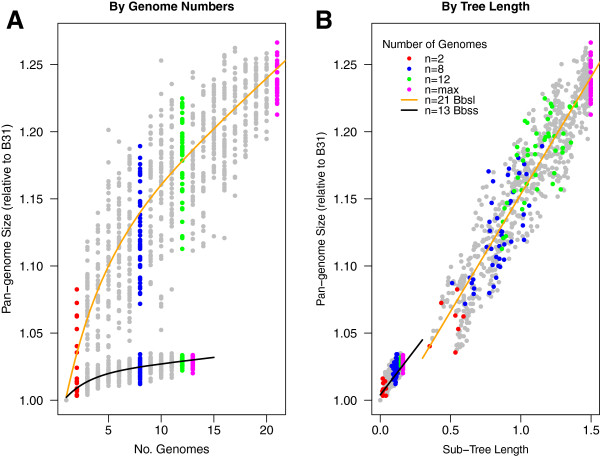
Intra- and inter-specific pan-genome sizes of B. burgdorferi s.l. expand linearly with phylogenetic diversity. Yet gene-acquisition rates in B. burgdorferi s.l. are among the lowest in bacterial pathogens, resulting in high genome stability and few lineage-specific genes. Genome adaptation of B. burgdorferi s.l. is driven predominantly by copy-number and sequence variations of lipoprotein genes. New genomic groups are likely to emerge if the current trend of B. burgdorferi s.l. population expansion continues.
莱姆病是由螺旋体细菌引起的,这些细菌来自博尔纳病螺旋体亚种(B. burgdorferi s.l.)复合群。为了重建博尔纳病螺旋体亚种的进化,并确定其人类毒力的基因组基础,我们比较了来自欧洲和美国的 23 种博尔纳病螺旋体亚种的基因组,包括博尔纳病亚种(B. burgdorferi s.s.,14 株)、阿费尔森病螺旋体(B. afzelii,2 株)、加氏病螺旋体(B. garinii,2 株)、“bavariensis”病螺旋体(B. "bavariensis",1 株)、斯氏病螺旋体(B. spielmanii,1 株)、瓦莱州病螺旋体(B. valaisiana,1 株)、比斯病螺旋体(B. bissettii,1 株)和“finlandensis”病螺旋体(B. "finlandensis",1 株)。
尽管存在重组,但使用全基因组单核苷酸多态性获得了稳健的博尔纳病亚种和博尔纳病螺旋体亚种的系统发育树。基于系统发育的泛基因组分析表明,种间基因获取率高于种内,表明适应性物种形成。强烈的正自然选择驱动脂蛋白的序列进化,包括染色体编码基因 0102 和 0404、cp26 编码的 ospC 和 b08 以及 lp54 编码的 dbpA、a07、a22、a33、a53、a65。计算机模拟预测,随着种群规模的增加,基因组群体的快速适应性辐射是可能的。
博尔纳病螺旋体亚种的种内和种间泛基因组大小与系统发育多样性呈线性扩展。然而,博尔纳病螺旋体亚种的基因获取率在细菌病原体中是最低的,导致基因组高度稳定,谱系特异性基因很少。博尔纳病螺旋体亚种的基因组适应主要由脂蛋白基因的拷贝数和序列变异驱动。如果当前博尔纳病螺旋体亚种种群扩张的趋势继续下去,新的基因组群体可能会出现。