Berglund Eva C, Lindqvist Carl Mårten, Hayat Shahina, Övernäs Elin, Henriksson Niklas, Nordlund Jessica, Wahlberg Per, Forestier Erik, Lönnerholm Gudmar, Syvänen Ann-Christine
Department of Medical Sciences, Molecular Medicine and Science for Life Laboratory, Uppsala University, Uppsala, Sweden.
BMC Genomics. 2013 Dec 5;14(1):856. doi: 10.1186/1471-2164-14-856.
Target enrichment and resequencing is a widely used approach for identification of cancer genes and genetic variants associated with diseases. Although cost effective compared to whole genome sequencing, analysis of many samples constitutes a significant cost, which could be reduced by pooling samples before capture. Another limitation to the number of cancer samples that can be analyzed is often the amount of available tumor DNA. We evaluated the performance of whole genome amplified DNA and the power to detect subclonal somatic single nucleotide variants in non-indexed pools of cancer samples using the HaloPlex technology for target enrichment and next generation sequencing.
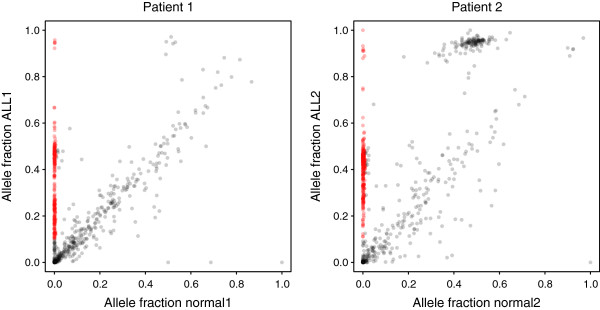
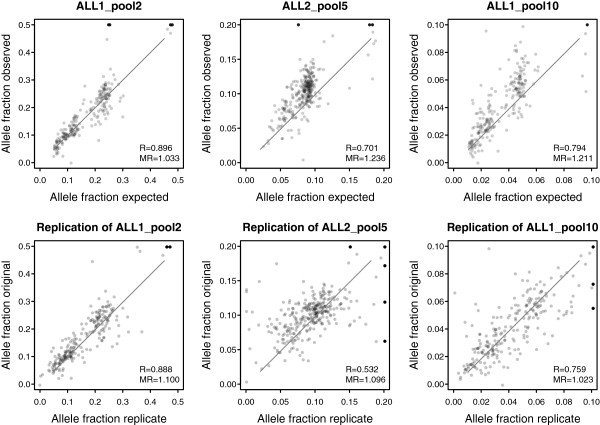
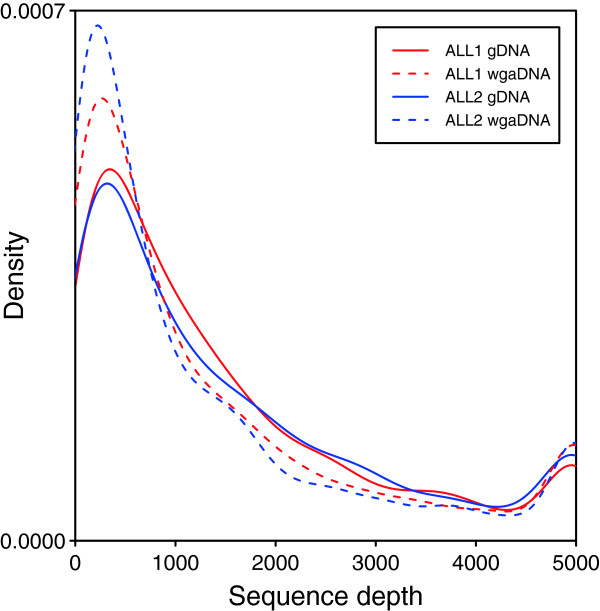
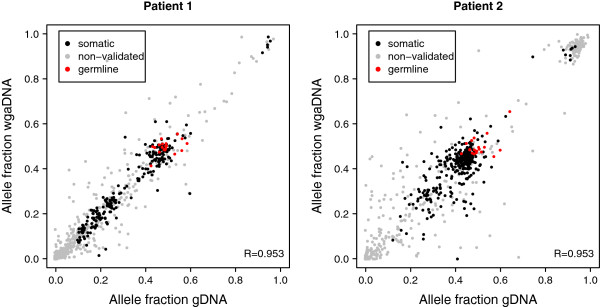
We captured a set of 1528 putative somatic single nucleotide variants and germline SNPs, which were identified by whole genome sequencing, with the HaloPlex technology and sequenced to a depth of 792-1752. We found that the allele fractions of the analyzed variants are well preserved during whole genome amplification and that capture specificity or variant calling is not affected. We detected a large majority of the known single nucleotide variants present uniquely in one sample with allele fractions as low as 0.1 in non-indexed pools of up to ten samples. We also identified and experimentally validated six novel variants in the samples included in the pools.
Our work demonstrates that whole genome amplified DNA can be used for target enrichment equally well as genomic DNA and that accurate variant detection is possible in non-indexed pools of cancer samples. These findings show that analysis of a large number of samples is feasible at low cost, even when only small amounts of DNA is available, and thereby significantly increases the chances of indentifying recurrent mutations in cancer samples.
靶向富集和重测序是一种广泛用于鉴定癌症基因和与疾病相关的遗传变异的方法。尽管与全基因组测序相比具有成本效益,但分析许多样本构成了一项重大成本,通过在捕获前合并样本可以降低这一成本。可分析的癌症样本数量的另一个限制通常是可用肿瘤DNA的量。我们使用HaloPlex技术进行靶向富集和下一代测序,评估了全基因组扩增DNA的性能以及在未索引的癌症样本池中检测亚克隆体细胞单核苷酸变异的能力。
我们利用HaloPlex技术捕获了一组通过全基因组测序鉴定的1528个推定的体细胞单核苷酸变异和种系单核苷酸多态性,并测序至792 - 1752的深度。我们发现,在全基因组扩增过程中,分析变异的等位基因分数得到了很好的保留,并且捕获特异性或变异检出不受影响。我们在多达十个样本的未索引样本池中检测到了绝大多数仅在一个样本中存在的已知单核苷酸变异,其等位基因分数低至0.1。我们还在样本池中包含的样本中鉴定并通过实验验证了六个新变异。
我们的工作表明,全基因组扩增DNA可与基因组DNA一样很好地用于靶向富集,并且在未索引的癌症样本池中可以进行准确的变异检测。这些发现表明,即使只有少量DNA可用,低成本分析大量样本也是可行的,从而显著增加了在癌症样本中鉴定复发突变的机会。