Kjellström Ulrika
Department of Ophthalmology, University of Lund, Lund, Sweden.
Mol Vis. 2014 Jan 7;20:89-104. eCollection 2014.
To investigate the genotype and phenotype in families with adenosine triphosphate-binding cassette, sub-family A, member 4 (ABCA4)-associated retinal degeneration.
Three families with at least one family member with known homozygous or compound heterozygote mutations in the ABCA4 gene were studied. The investigations included full field electroretinography (ff-ERG), multifocal ERG (mERG), Goldmann visual fields, optical coherence tomography (OCT), and standard ophthalmological examination. Microarray (Asper) was used for ABCA4 genotyping.
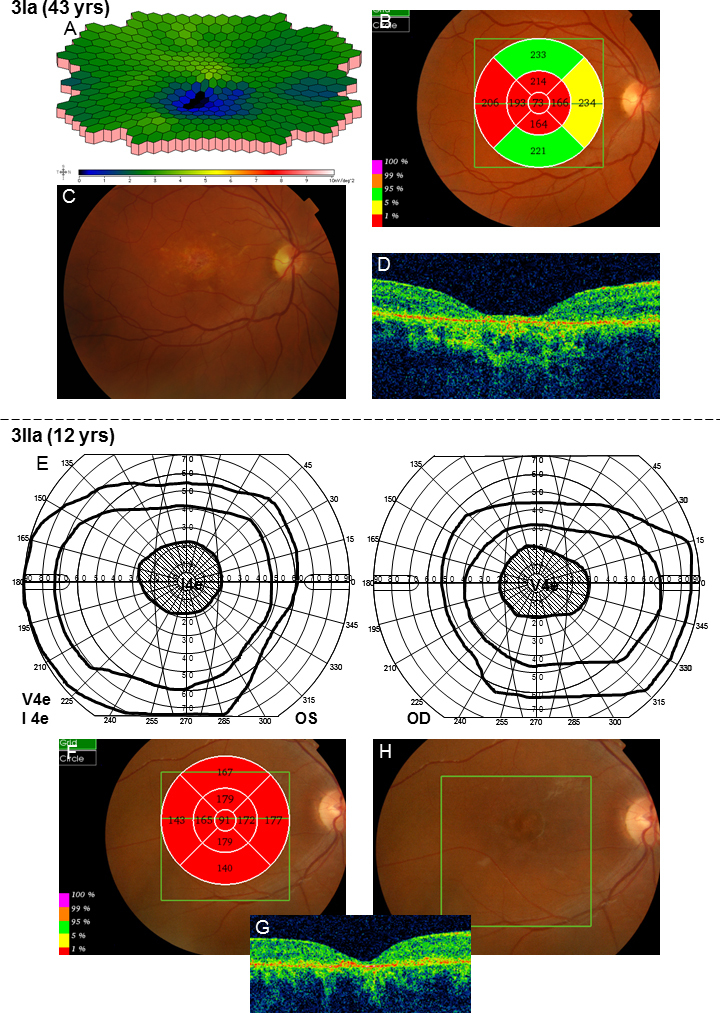
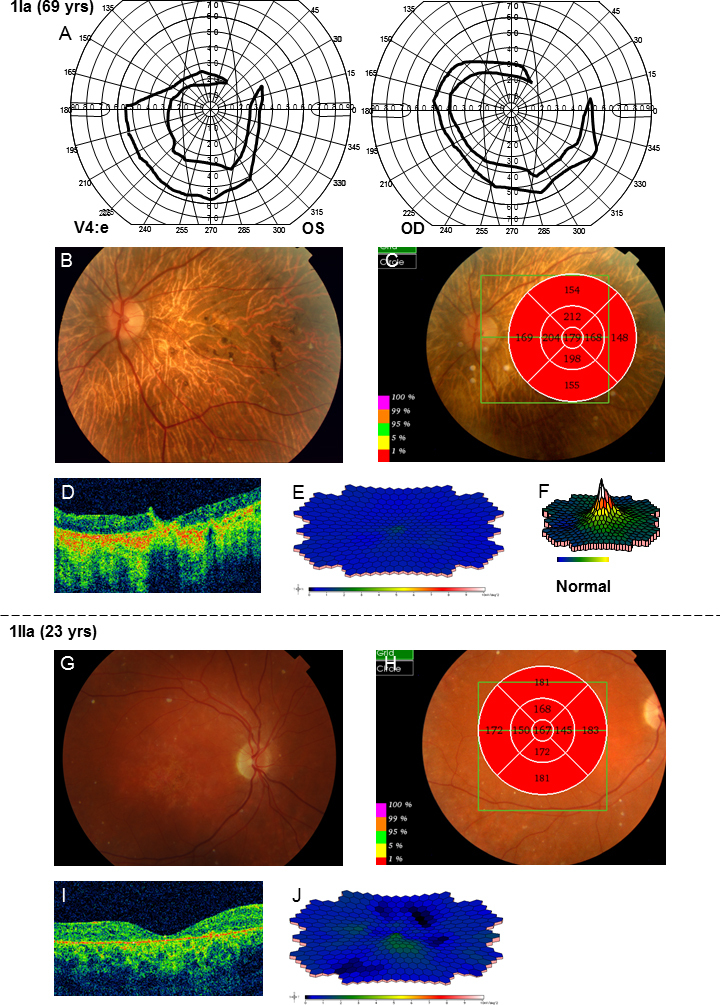
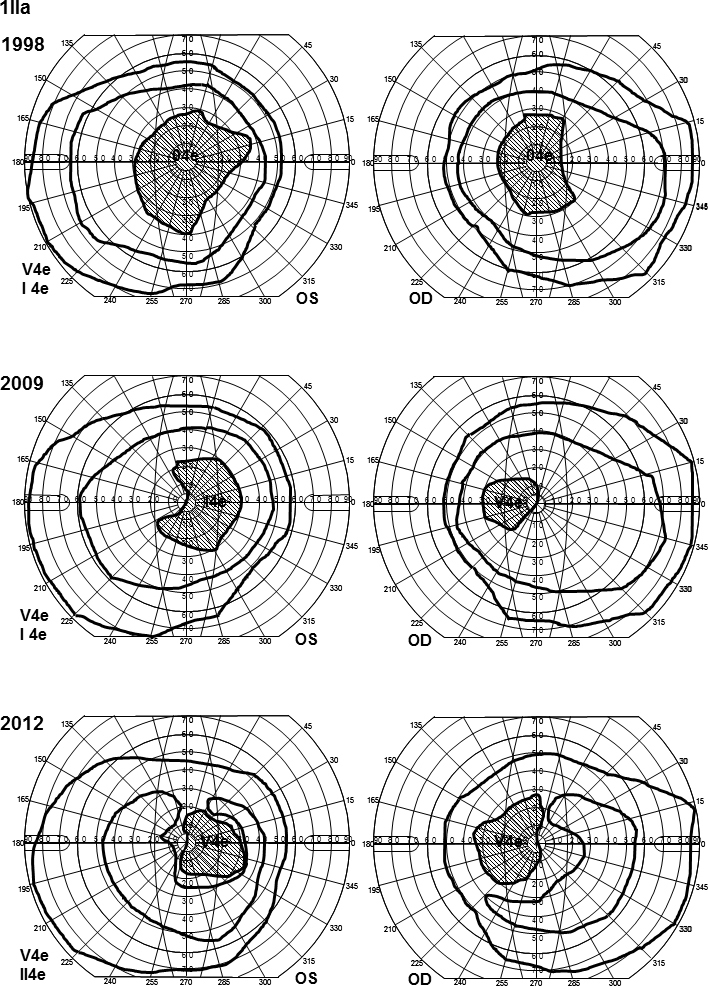
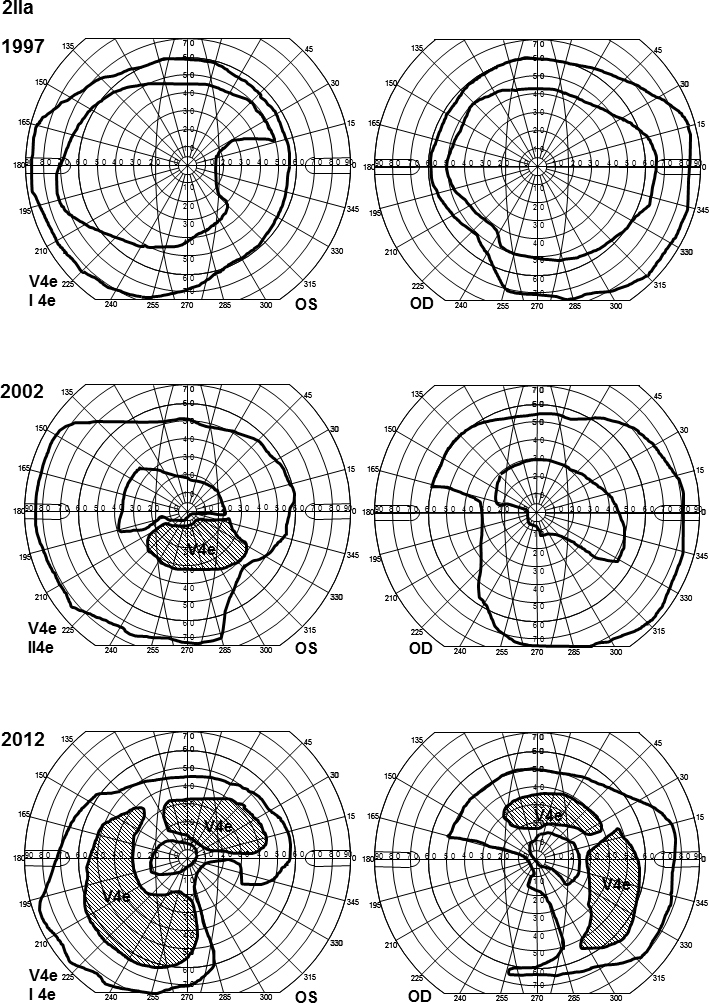
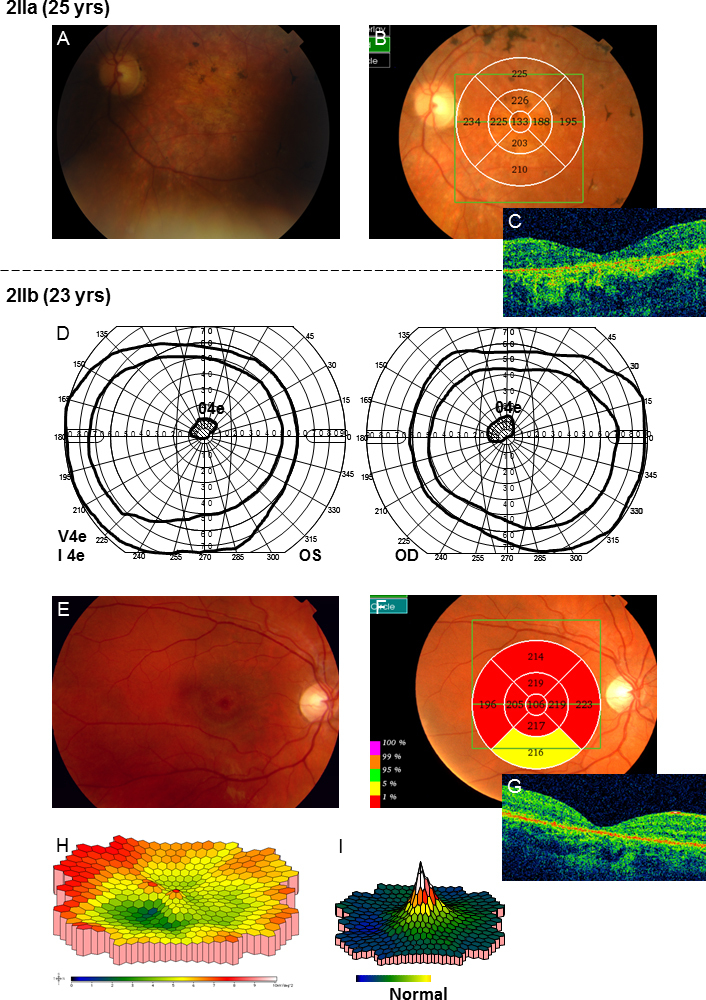
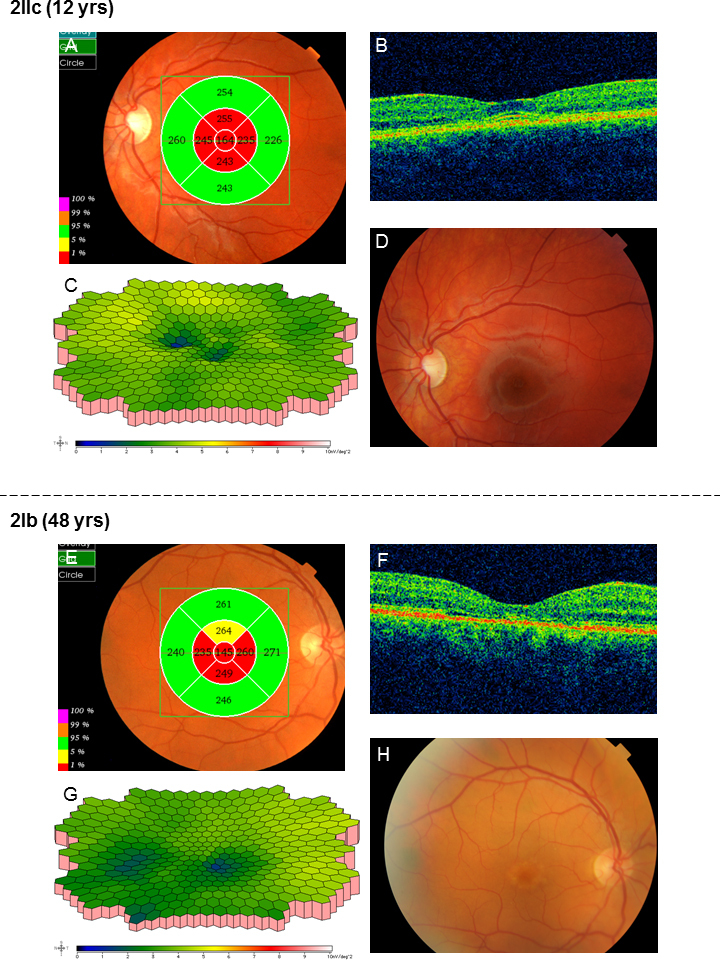
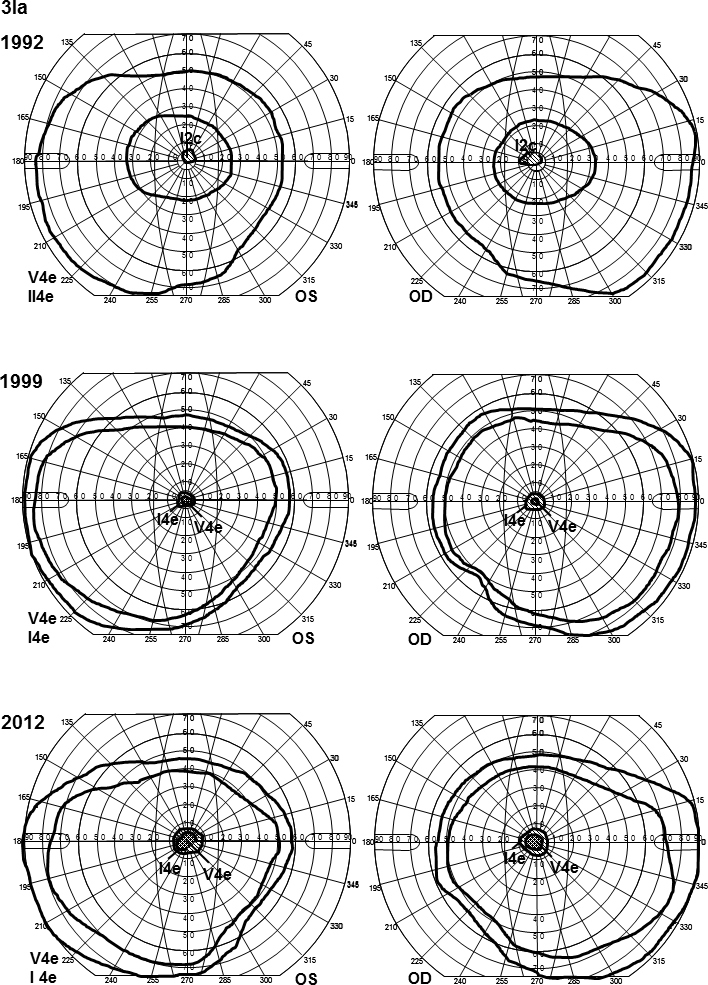
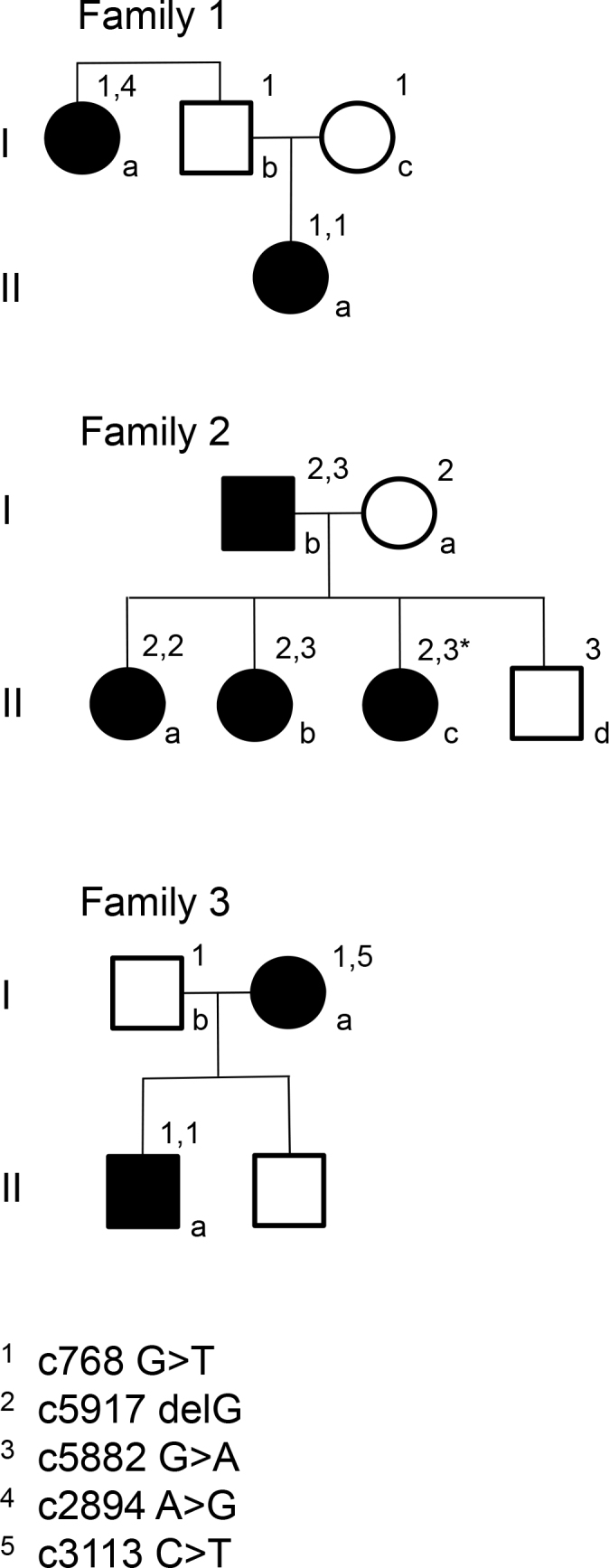
In family 1, the proband (age 23) was homozygote for the c768 G>T mutation. She was diagnosed with cone rod dystrophy (CRD) while her aunt (age 69) was compound heterozygote for the c768 G>T and c2894 A>G mutations and had autosomal recessive retinitis pigmentosa (arRP). The father (age 61) and the mother (age 60) of the proband were asymptomatic carriers of the c768 G>T mutation. In family 2, the proband (age 25) was homozygote for the c5917del. She was diagnosed with CRD. Her father and two sisters were compound heterozygote for the c5917del and c5882 G>A mutations. The eldest sister (age 23) suffered from Stargardt disease (STGD) while the youngest sister (age 12) and their father (age 48) had no visual complaints. Anyhow, their ERG measurements indicated changes corresponding to STGD. The mother (age 42), (heterozygote for the c5917 delG mutation) and the youngest child (age 9; heterozygote for the c5882 G>A mutation) had a normal phenotype. In family 3, the proband (age 43) was compound heterozygote for c768 G>T and c3113 C>T and had been diagnosed with STGD. Her son (age 12), who was homozygote for the c768 G>T mutation, had wider scotomas with earlier onset (age 6), ff-ERG cone responses in the lower range of normality, and reduced mERG. At the moment, he is classified as having STGD but may progress to CRD. The father (age 45) was asymptomatic and heterozygote for the c768 G>T mutation. The patients with progressive disorders (CRD or arRP) had prolonged implicit times for the 30 Hz flicker ff-ERG and the mERG. All patients with two mutations in the ABCA4 gene demonstrated attenuation of retinal thickness on the OCT macular map.
This study confirms that ABCA4 mutations lead to a spectrum of retinal degenerations ranging from STGD to CRD or arRP. At the time of diagnosis, it is not possible to predict the severity of the condition only from genotyping. Our results suggest that prolongation of implicit times for the ff-ERG and/or mERG seem to be associated with progressive conditions such as CRD and arRP. Since ABCA4 mutations are common in the general population, different family members can harbor various combinations of mutations resulting in diverse phenotype and prognosis in the same family, further emphasizing the importance of a combination of genetic and electrophysiological tests at the first visit and follow-up.
研究三磷酸腺苷结合盒转运体A亚家族成员4(ABCA4)相关视网膜变性家系的基因型和表型。
研究了三个家系,每个家系中至少有一名家庭成员在ABCA4基因中存在已知的纯合或复合杂合突变。研究内容包括全视野视网膜电图(ff-ERG)、多焦视网膜电图(mERG)、Goldmann视野检查、光学相干断层扫描(OCT)和标准眼科检查。采用微阵列(Asper)进行ABCA4基因分型。
在家族1中,先证者(23岁)为c768 G>T突变的纯合子,被诊断为锥杆营养不良(CRD),而她的姑姑(69岁)为c768 G>T和c2894 A>G突变的复合杂合子,患有常染色体隐性遗传性视网膜色素变性(arRP)。先证者的父亲(61岁)和母亲(60岁)是c768 G>T突变的无症状携带者。在家族2中,先证者(25岁)为c5917del突变的纯合子,被诊断为CRD。她的父亲和两个姐妹是c5917del和c5882 G>A突变的复合杂合子。大姐(23岁)患有Stargardt病(STGD),而小妹(12岁)和她们的父亲(48岁)没有视觉症状。然而,他们的视网膜电图测量结果显示出与STGD相符的变化。母亲(42岁,c5917 delG突变杂合子)和最小的孩子(9岁,c5882 G>A突变杂合子)表型正常。在家族3中,先证者(43岁)是c768 G>T和c3113 C>T突变的复合杂合子,被诊断为STGD。她的儿子(12岁)是c768 G>T突变的纯合子,有更广泛的暗点且发病更早(6岁),ff-ERG锥体反应处于正常范围下限,mERG降低。目前,他被归类为患有STGD,但可能会发展为CRD。父亲(45岁)无症状,是c768 G>T突变的杂合子。患有进行性疾病(CRD或arRP)的患者30 Hz闪烁ff-ERG和mERG的隐含时间延长。所有ABCA4基因有两个突变的患者在OCT黄斑图上均显示视网膜厚度变薄。
本研究证实ABCA4突变可导致一系列视网膜变性,从STGD到CRD或arRP。在诊断时,仅通过基因分型无法预测病情的严重程度。我们的结果表明,ff-ERG和/或mERG隐含时间的延长似乎与CRD和arRP等进行性疾病相关。由于ABCA4突变在普通人群中很常见,不同家庭成员可能携带不同的突变组合,导致同一家系中出现不同的表型和预后,这进一步强调了在首次就诊和随访时进行基因和电生理检查相结合的重要性。