Byrne Lisa, Elson Richard, Dallman Timothy J, Perry Neil, Ashton Philip, Wain John, Adak Goutam K, Grant Kathie A, Jenkins Claire
Gastrointestinal Emerging and Zoonotic Infections Department, Health Protection Services, Public Health England, Centre for Infectious Disease Surveillance and Control, London, United Kingdom.
Gastrointestinal Bacteria Reference Unit, Microbiology Services, GBRU, Public Health England, Centre for Infectious Disease Surveillance and Control, London, United Kingdom.
PLoS One. 2014 Jan 17;9(1):e85901. doi: 10.1371/journal.pone.0085901. eCollection 2014.
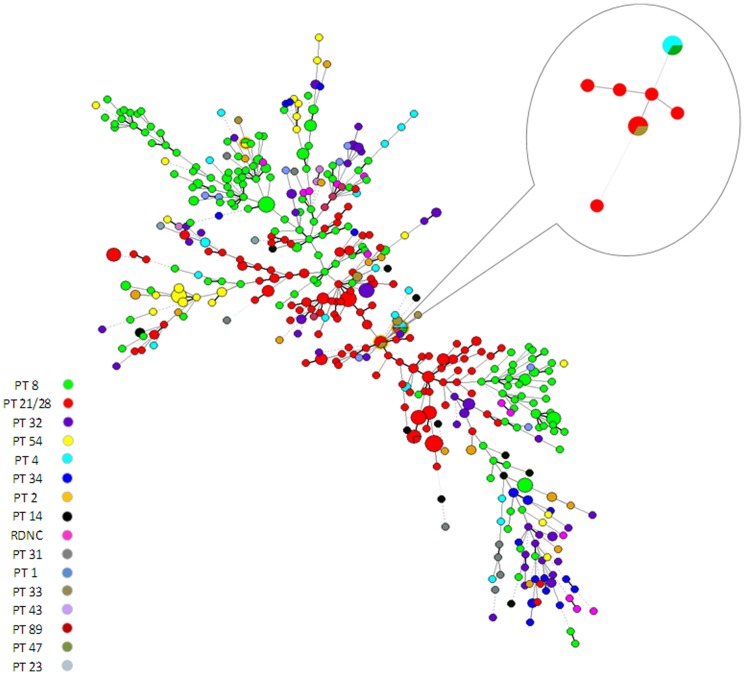
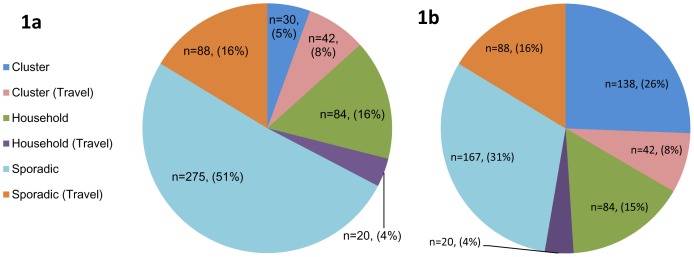
Multilocus variable number tandem repeat analysis (MLVA) provides microbiological support for investigations of clusters of cases of infection with Shiga toxin-producing E. coli (STEC) O157. All confirmed STEC O157 isolated in England and submitted to the Gastrointestinal Bacteria Reference Unit (GBRU) during a six month period were typed using MLVA, with the aim of assessing the impact of this approach on epidemiological investigations. Of 539 cases investigated, 341 (76%) had unique (>2 single locus variants) MLVA profiles, 12% of profiles occurred more than once due to known household transmission and 12% of profiles occurred as part of 41 clusters, 21 of which were previously identified through routine public health investigation of cases. The remaining 20 clusters were not previously detected and STEC enhanced surveillance data for associated cases were retrospectively reviewed for epidemiological links including shared exposures, geography and/or time. Additional evidence of a link between cases was found in twelve clusters. Compared to phage typing, the number of sporadic cases was reduced from 69% to 41% and the diversity index for MLVA was 0.996 versus 0.782 for phage typing. Using MLVA generates more data on the spatial and temporal dispersion of cases, better defining the epidemiology of STEC infection than phage typing. The increased detection of clusters through MLVA typing highlights the challenges to health protection practices, providing a forerunner to the advent of whole genome sequencing as a diagnostic tool.
多位点可变数目串联重复序列分析(MLVA)为产志贺毒素大肠杆菌(STEC)O157感染病例群的调查提供了微生物学支持。在六个月期间,对所有在英格兰分离并提交给胃肠道细菌参考单位(GBRU)的确诊STEC O157菌株进行了MLVA分型,目的是评估这种方法对流行病学调查的影响。在调查的539例病例中,341例(76%)具有独特的(>2个单基因座变异)MLVA图谱,12%的图谱因已知的家庭传播而出现不止一次,12%的图谱作为41个簇的一部分出现,其中21个簇先前是通过对病例的常规公共卫生调查确定的。其余20个簇以前未被检测到,对相关病例的STEC强化监测数据进行了回顾性审查,以寻找包括共同暴露、地理位置和/或时间在内的流行病学联系。在12个簇中发现了病例之间存在联系的更多证据。与噬菌体分型相比,散发病例的数量从69%减少到41%,MLVA的多样性指数为0.996,而噬菌体分型为0.782。使用MLVA可以生成更多关于病例时空分布的数据,比噬菌体分型能更好地界定STEC感染的流行病学特征。通过MLVA分型增加对簇的检测凸显了卫生防护措施面临的挑战,为全基因组测序作为一种诊断工具的出现提供了先兆。