Department of Pathology, University of Veterinary Medicine Hannover, Hannover, Germany ; Center for Systems Neuroscience, University of Veterinary Medicine Hannover, Hannover, Germany.
Department of Non-Clinical Drug Safety, Boehringer Ingelheim Pharma GmbH&Co KG, Biberach (Riß), Germany.
PLoS One. 2014 Jan 27;9(1):e86643. doi: 10.1371/journal.pone.0086643. eCollection 2014.
Multiple microarray analyses of multiple sclerosis (MS) and its experimental models have been published in the last years.
Meta-analyses integrate the information from multiple studies and are suggested to be a powerful approach in detecting highly relevant and commonly affected pathways.
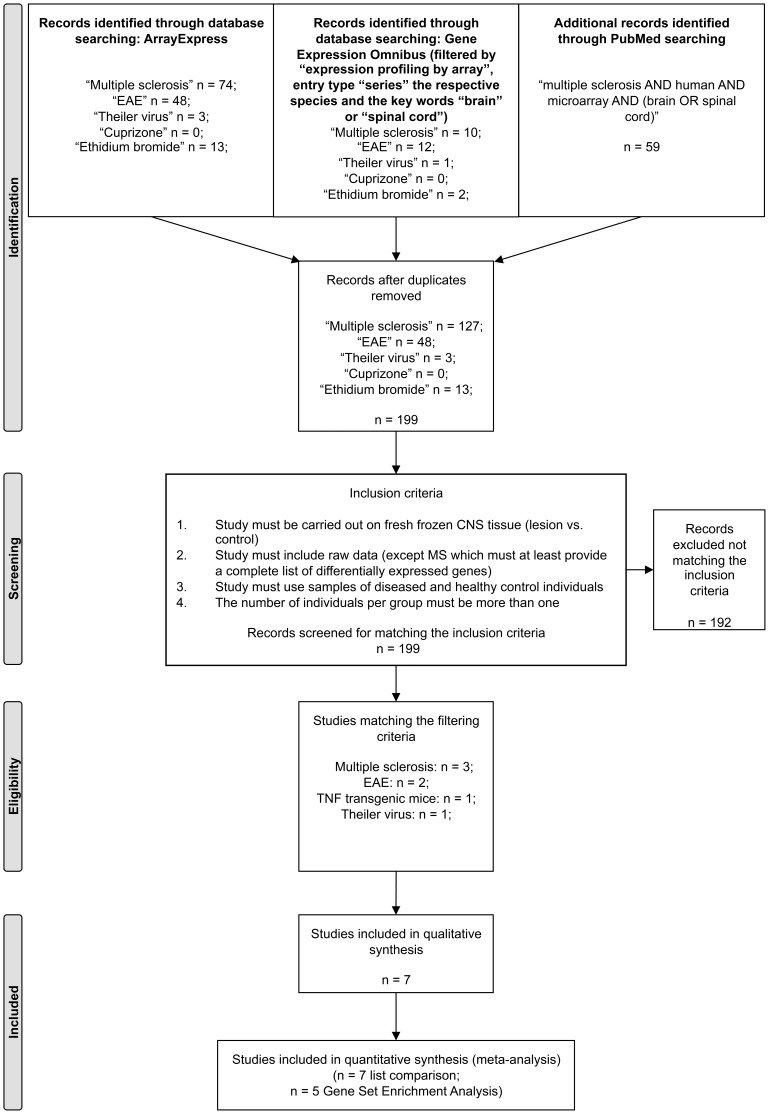
ArrayExpress, Gene Expression Omnibus and PubMed databases were screened for microarray gene expression profiling studies of MS and its experimental animal models.
Studies comparing central nervous system (CNS) samples of diseased versus healthy individuals with n >1 per group and publically available raw data were selected.
Included conditions for re-analysis of differentially expressed genes (DEGs) were MS, myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis (EAE) in rats, proteolipid protein-induced EAE in mice, Theiler's murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD), and a transgenic tumor necrosis factor-overexpressing mouse model (TNFtg). Since solely a single MS raw data set fulfilled the inclusion criteria, a merged list containing the DEGs from two MS-studies was additionally included. Cross-study analysis was performed employing list comparisons of DEGs and alternatively Gene Set Enrichment Analysis (GSEA).
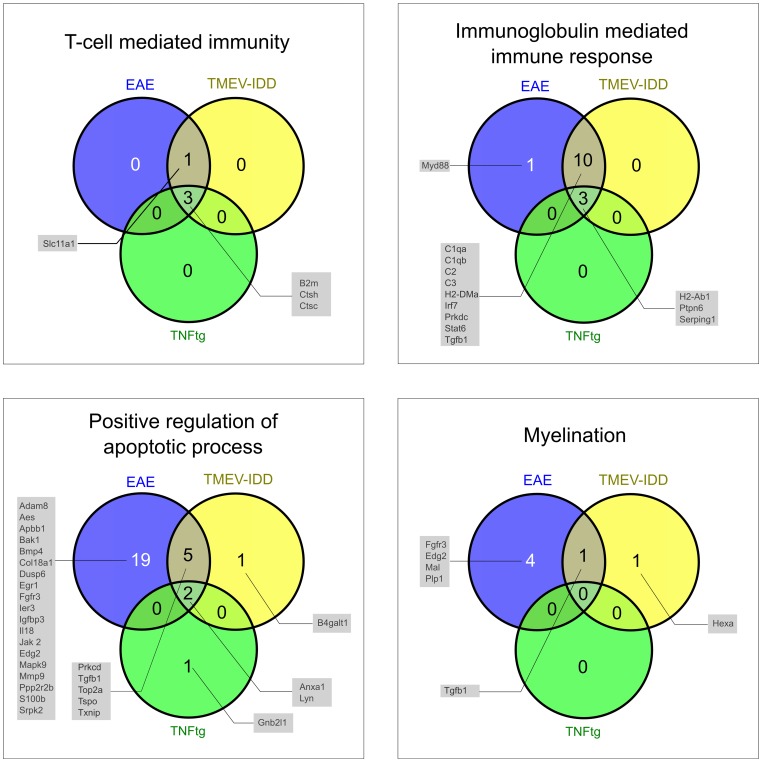
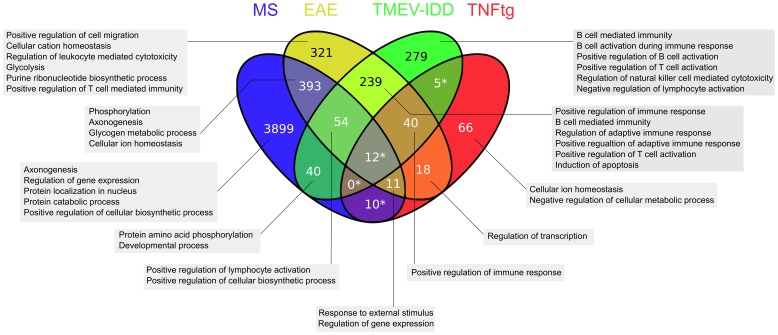
The intersection of DEGs in MS, EAE, TMEV-IDD, and TNFtg contained 12 genes related to macrophage functions. The intersection of EAE, TMEV-IDD and TNFtg comprised 40 DEGs, functionally related to positive regulation of immune response. Over and above, GSEA identified substantially more differentially regulated pathways including coagulation and JAK/STAT-signaling.
A meta-analysis based on a simple comparison of DEGs is over-conservative. In contrast, the more experimental GSEA approach identified both, a priori anticipated as well as promising new candidate pathways.
多发性硬化症(MS)及其实验模型的多项微阵列分析已在过去几年中发表。
荟萃分析整合了来自多个研究的信息,被认为是检测高度相关和普遍受影响途径的强大方法。
ArrayExpress、基因表达综合数据库和 PubMed 数据库被筛选用于 MS 及其实验动物模型的微阵列基因表达谱研究。
选择了比较中枢神经系统(CNS)疾病与健康个体样本的研究,每组 n>1,并且具有公开的原始数据。
对差异表达基因(DEGs)进行重新分析的纳入条件为 MS、大鼠髓鞘少突胶质细胞糖蛋白诱导的实验性自身免疫性脑脊髓炎(EAE)、小鼠蛋白脂质蛋白诱导的 EAE、Theiler 鼠脑炎病毒诱导的脱髓鞘疾病(TMEV-IDD)和转肿瘤坏死因子过表达小鼠模型(TNFtg)。由于仅有一个 MS 原始数据集符合纳入标准,因此还额外纳入了一个包含两个 MS 研究的 DEGs 的合并列表。采用 DEGs 的列表比较和替代基因集富集分析(GSEA)进行跨研究分析。
MS、EAE、TMEV-IDD 和 TNFtg 的 DEGs 交集包含 12 个与巨噬细胞功能相关的基因。EAE、TMEV-IDD 和 TNFtg 的交集包含 40 个 DEGs,与免疫反应的正调节功能相关。此外,GSEA 还鉴定出了更多差异调节的途径,包括凝血和 JAK/STAT 信号通路。
基于 DEGs 简单比较的荟萃分析过于保守。相比之下,更具实验性的 GSEA 方法不仅识别了预期的途径,还识别了有前途的新候选途径。