Institute for Genome Sciences, Department of Microbiology and Immunology, University of Maryland School of Medicine, 801 W, Baltimore Street, Baltimore, MD 21201, USA.
Microbiome. 2014 Feb 24;2(1):6. doi: 10.1186/2049-2618-2-6.
To take advantage of affordable high-throughput next-generation sequencing technologies to characterize microbial community composition often requires the development of improved methods to overcome technical limitations inherent to the sequencing platforms. Sequencing low sequence diversity libraries such as 16S rRNA amplicons has been problematic on the Illumina MiSeq platform and often generates sequences of suboptimal quality.
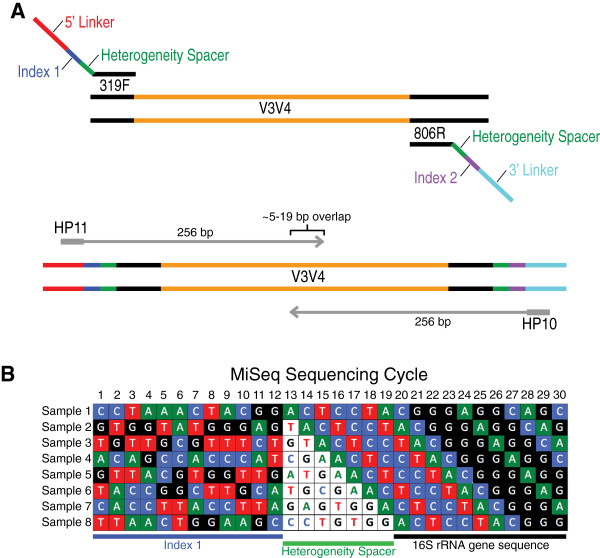
Here we present an improved dual-indexing amplification and sequencing approach to assess the composition of microbial communities from clinical samples using the V3-V4 region of the 16S rRNA gene on the Illumina MiSeq platform. We introduced a 0 to 7 bp "heterogeneity spacer" to the index sequence that allows an equal proportion of samples to be sequenced out of phase.
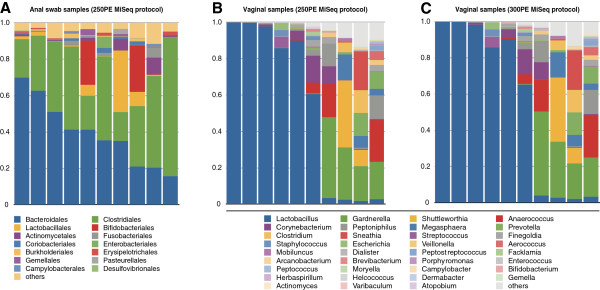
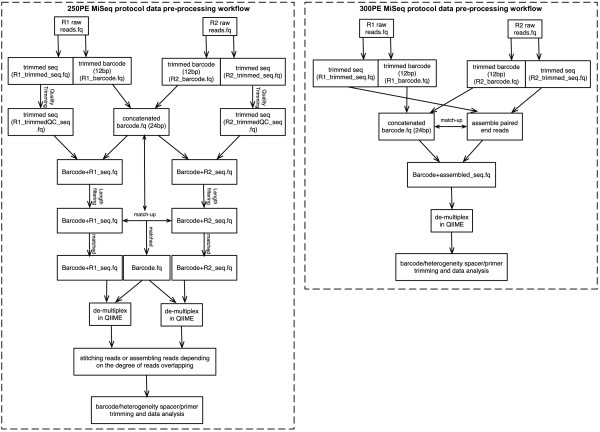
Our approach yields high quality sequence data from 16S rRNA gene amplicons using both 250 bp and 300 bp paired-end MiSeq protocols and provides a flexible and cost-effective sequencing option.
为了利用价格合理的高通量下一代测序技术来描述微生物群落的组成,通常需要开发改进的方法来克服测序平台固有的技术限制。在 Illumina MiSeq 平台上对 16S rRNA 扩增子等低序列多样性文库进行测序一直存在问题,并且经常会生成质量不佳的序列。
在这里,我们提出了一种改进的双索引扩增和测序方法,用于使用 Illumina MiSeq 平台上的 16S rRNA 基因的 V3-V4 区域评估临床样本中微生物群落的组成。我们在索引序列中引入了 0 到 7 bp 的“异质间隔区”,允许等比例的样本进行异相测序。
我们的方法使用 250 bp 和 300 bp 配对末端 MiSeq 方案从 16S rRNA 基因扩增子中获得高质量的序列数据,并提供了一种灵活且具有成本效益的测序选择。