Kosciolek Tomasz, Jones David T
Bioinformatics Group, Department of Computer Science, University College London, London, United Kingdom; Institute of Structural and Molecular Biology, University College London, London, United Kingdom.
PLoS One. 2014 Mar 17;9(3):e92197. doi: 10.1371/journal.pone.0092197. eCollection 2014.
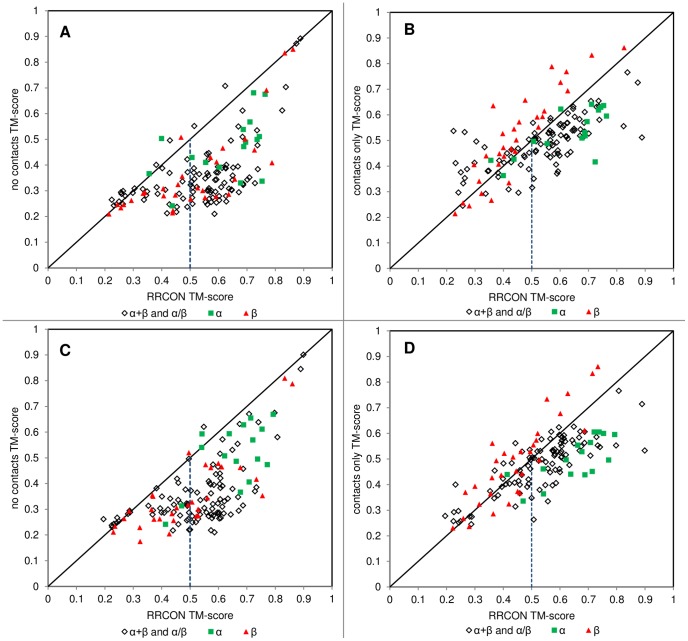
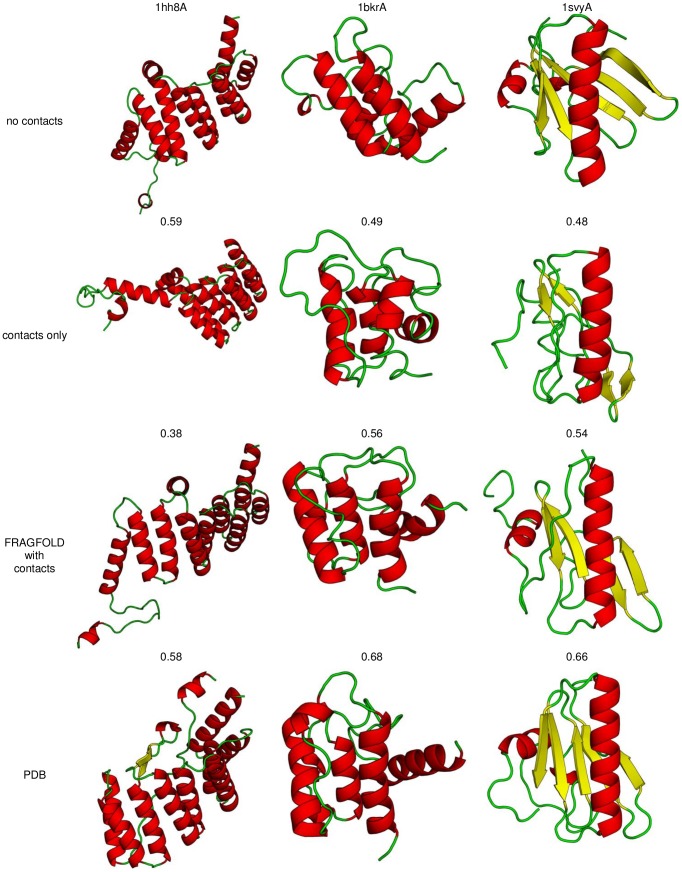
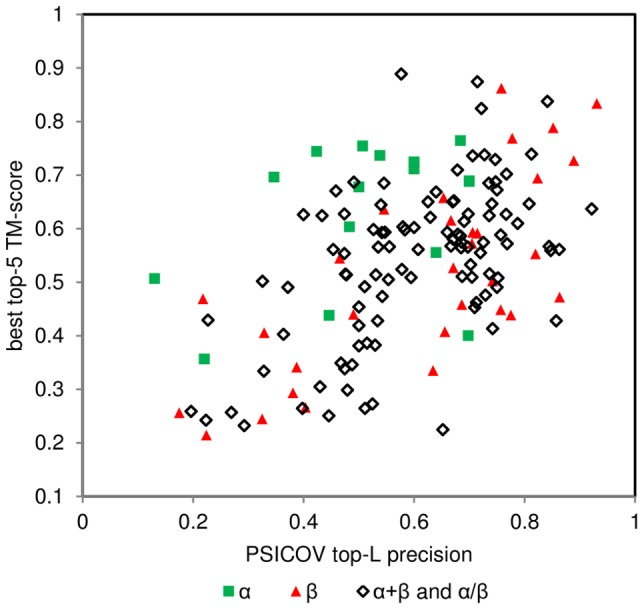
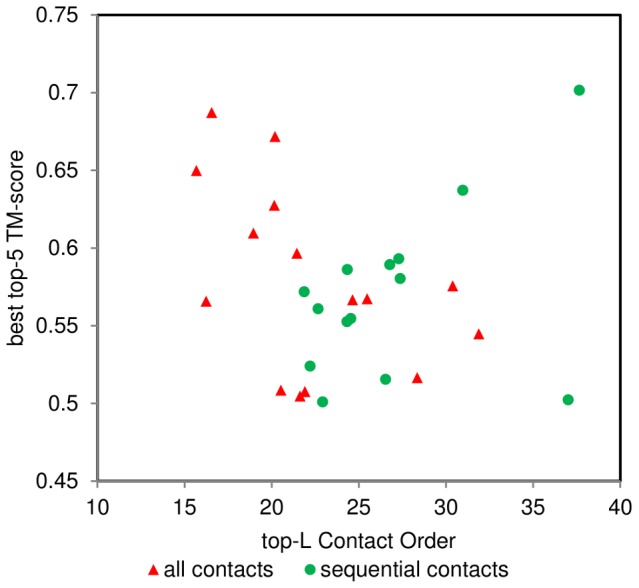
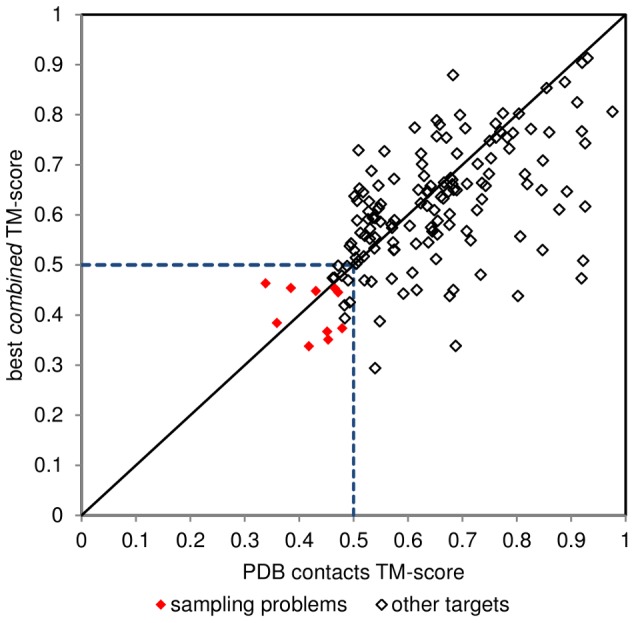
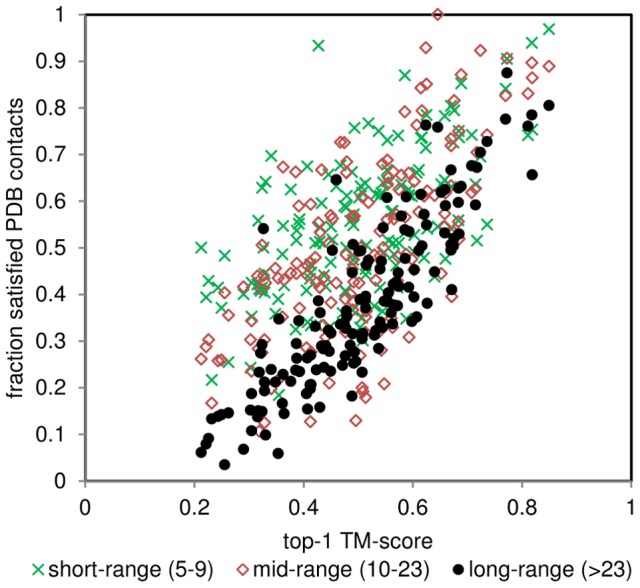
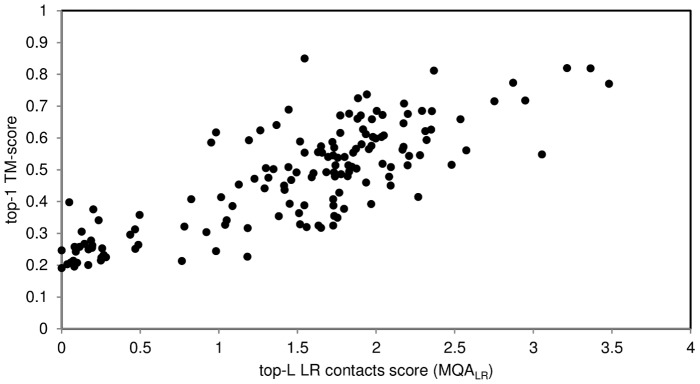
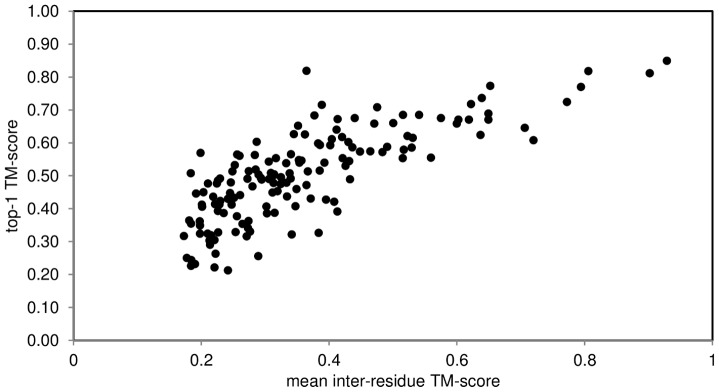
The advent of high accuracy residue-residue intra-protein contact prediction methods enabled a significant boost in the quality of de novo structure predictions. Here, we investigate the potential benefits of combining a well-established fragment-based folding algorithm--FRAGFOLD, with PSICOV, a contact prediction method which uses sparse inverse covariance estimation to identify co-varying sites in multiple sequence alignments. Using a comprehensive set of 150 diverse globular target proteins, up to 266 amino acids in length, we are able to address the effectiveness and some limitations of such approaches to globular proteins in practice. Overall we find that using fragment assembly with both statistical potentials and predicted contacts is significantly better than either statistical potentials or contacts alone. Results show up to nearly 80% of correct predictions (TM-score ≥0.5) within analysed dataset and a mean TM-score of 0.54. Unsuccessful modelling cases emerged either from conformational sampling problems, or insufficient contact prediction accuracy. Nevertheless, a strong dependency of the quality of final models on the fraction of satisfied predicted long-range contacts was observed. This not only highlights the importance of these contacts on determining the protein fold, but also (combined with other ensemble-derived qualities) provides a powerful guide as to the choice of correct models and the global quality of the selected model. A proposed quality assessment scoring function achieves 0.93 precision and 0.77 recall for the discrimination of correct folds on our dataset of decoys. These findings suggest the approach is well-suited for blind predictions on a variety of globular proteins of unknown 3D structure, provided that enough homologous sequences are available to construct a large and accurate multiple sequence alignment for the initial contact prediction step.
高精度蛋白质内残基-残基接触预测方法的出现,使得从头结构预测的质量得到了显著提升。在此,我们研究了将一种成熟的基于片段的折叠算法——FRAGFOLD与PSICOV相结合的潜在益处,PSICOV是一种接触预测方法,它使用稀疏逆协方差估计来识别多序列比对中的共变位点。使用一组包含150种不同的球状目标蛋白(长度可达266个氨基酸)的综合数据集,我们能够在实践中探讨此类方法对球状蛋白的有效性及一些局限性。总体而言,我们发现结合使用具有统计势和预测接触的片段组装方法,比单独使用统计势或接触要好得多。结果显示,在分析的数据集中,高达近80%的预测是正确的(TM分数≥0.5),平均TM分数为0.54。建模失败的情况要么源于构象采样问题,要么源于接触预测精度不足。然而,我们观察到最终模型的质量强烈依赖于满足预测的长程接触的比例。这不仅突出了这些接触在确定蛋白质折叠方面的重要性,而且(与其他源自集成的质量相结合)为正确模型的选择和所选模型的整体质量提供了有力指导。对于我们的诱饵数据集上正确折叠的判别,一种提出的质量评估评分函数实现了0.93的精度和0.77的召回率。这些发现表明,该方法非常适合对各种未知三维结构的球状蛋白进行盲预测,前提是有足够的同源序列可用于构建用于初始接触预测步骤的大型且准确的多序列比对。