Qu Jun, Young Rebeccah, Page Brian J, Shen Xiaomeng, Tata Nazneen, Li Jun, Duan Xiaotao, Fallavollita James A, Canty John M
Department of Pharmaceutical Sciences, ‡Department of Biochemistry, §Department of Medicine, ∥Department of Physiology and Biophysics, ⊥The Center for Research in Cardiovascular Medicine, and #Center for Excellence in Bioinformatics and Life Sciences, State University of New York at Buffalo , Buffalo, New York 14214, United States.
J Proteome Res. 2014 May 2;13(5):2571-84. doi: 10.1021/pr5000472. Epub 2014 Apr 4.
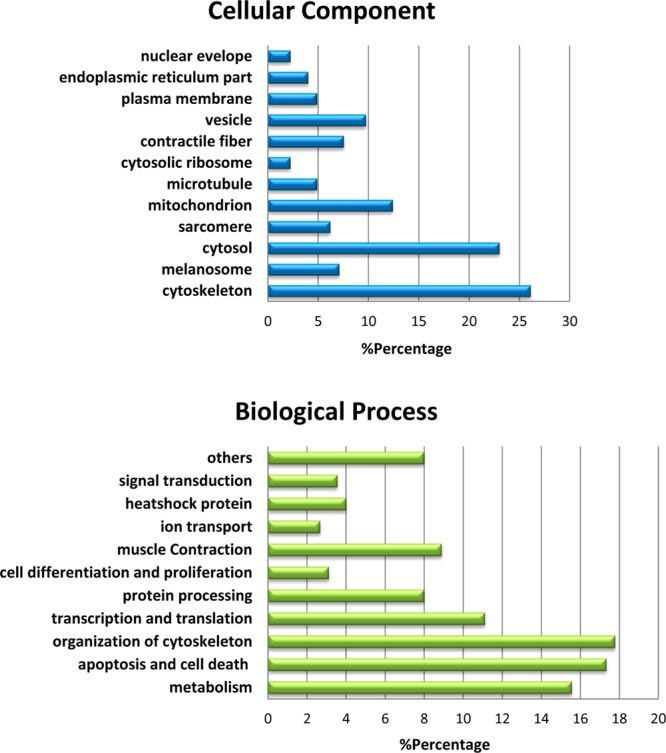
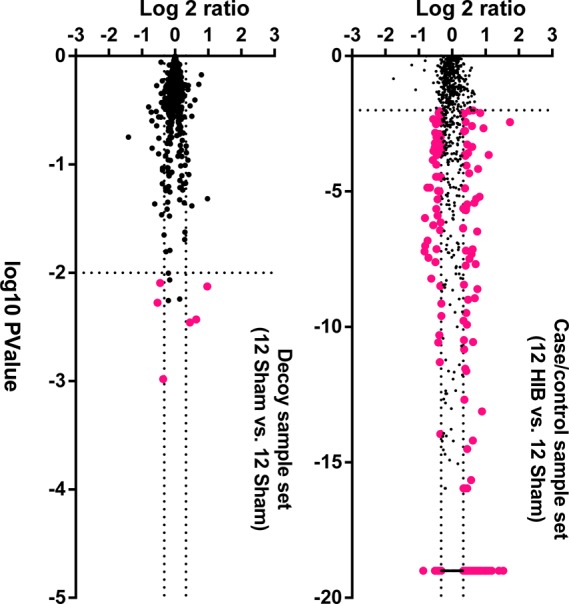
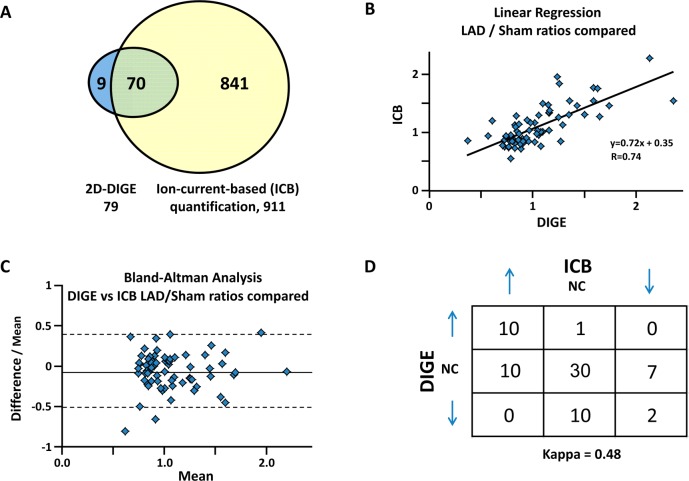
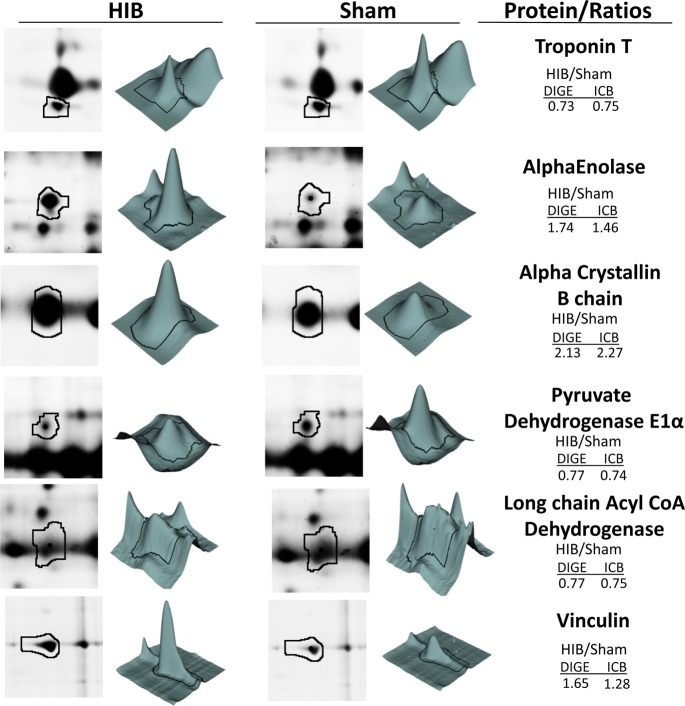
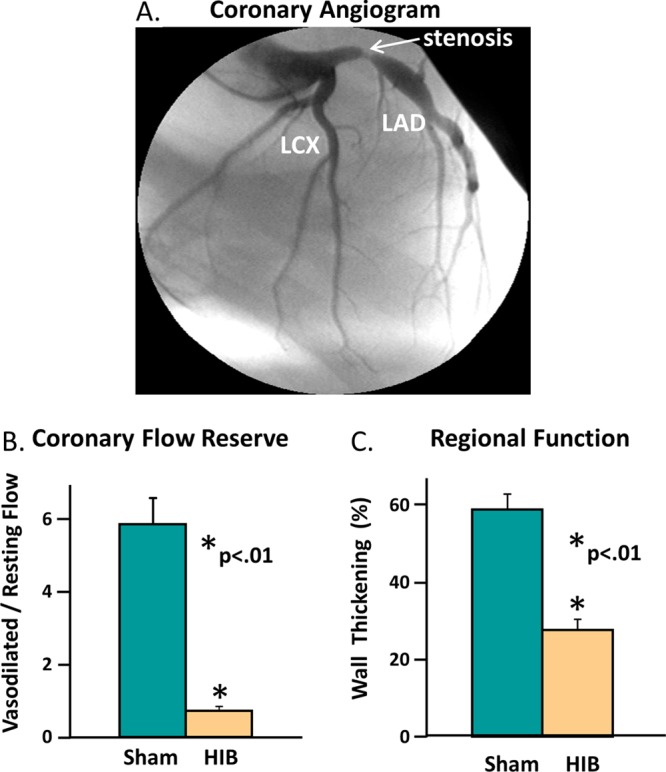
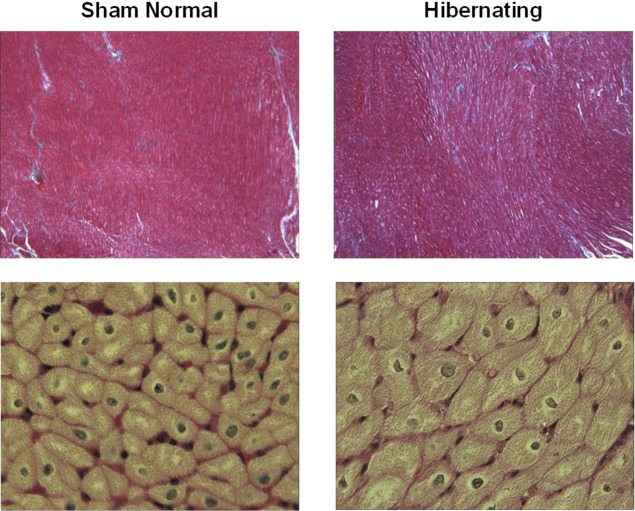
Hibernating myocardium is an adaptive response to repetitive myocardial ischemia that is clinically common, but the mechanism of adaptation is poorly understood. Here we compared the proteomes of hibernating versus normal myocardium in a porcine model with 24 biological replicates. Using the ion-current-based proteomic strategy optimized in this study to expand upon previous proteomic work, we identified differentially expressed proteins in new molecular pathways of cardiovascular interest. The methodological strategy includes efficient extraction with detergent cocktail; precipitation/digestion procedure with high, quantitative peptide recovery; reproducible nano-LC/MS analysis on a long, heated column packed with small particles; and quantification based on ion-current peak areas. Under the optimized conditions, high efficiency and reproducibility were achieved for each step, which enabled a reliable comparison of 24 the myocardial samples. To achieve confident discovery of differentially regulated proteins in hibernating myocardium, we used highly stringent criteria to define "quantifiable proteins". These included the filtering criteria of low peptide FDR and S/N > 10 for peptide ion currents, and each protein was quantified independently from ≥2 distinct peptides. For a broad methodological validation, the quantitative results were compared with a parallel, well-validated 2D-DIGE analysis of the same model. Excellent agreement between the two orthogonal methods was observed (R = 0.74), and the ion-current-based method quantified almost one order of magnitude more proteins. In hibernating myocardium, 225 significantly altered proteins were discovered with a low false-discovery rate (∼3%). These proteins are involved in biological processes including metabolism, apoptosis, stress response, contraction, cytoskeleton, transcription, and translation. This provides compelling evidence that hibernating myocardium adapts to chronic ischemia. The major metabolic mechanisms include a down-regulation of mitochondrial respiration and an increase in glycolysis. Meanwhile, cardioprotective and cytoskeletal proteins are increased, while cardiomyocyte contractile proteins are reduced. These intrinsic adaptations to regional ischemia maintain long-term cardiomyocyte viability at the expense of contractile function.
冬眠心肌是对反复心肌缺血的一种适应性反应,临床上较为常见,但对其适应机制了解甚少。在此,我们在猪模型中比较了冬眠心肌与正常心肌的蛋白质组,有24个生物学重复样本。利用本研究中优化的基于离子流的蛋白质组学策略,在先前蛋白质组学工作的基础上进行拓展,我们在心血管领域新的分子途径中鉴定出差异表达蛋白。该方法策略包括用去污剂混合物进行高效提取;采用具有高定量肽回收率的沉淀/消化程序;在填充有小颗粒的长加热柱上进行可重复的纳升液相色谱/质谱分析;以及基于离子流峰面积进行定量。在优化条件下,每个步骤都实现了高效率和可重复性,从而能够对24个心肌样本进行可靠比较。为了在冬眠心肌中可靠地发现差异调节蛋白,我们使用了高度严格的标准来定义“可定量蛋白”。这些标准包括低肽错误发现率(FDR)和肽离子流的信噪比>10的筛选标准,并且每个蛋白质独立地由≥2个不同的肽进行定量。为了进行广泛的方法学验证,将定量结果与同一模型的并行且经过充分验证的二维差异凝胶电泳(2D-DIGE)分析进行比较。观察到两种正交方法之间具有极好的一致性(R = 0.74),并且基于离子流的方法定量的蛋白质几乎多了一个数量级。在冬眠心肌中,发现了225种显著改变的蛋白,错误发现率较低(约3%)。这些蛋白质参与包括代谢、凋亡、应激反应、收缩、细胞骨架、转录和翻译等生物学过程。这提供了令人信服的证据,表明冬眠心肌适应慢性缺血。主要的代谢机制包括线粒体呼吸下调和糖酵解增加。同时,心脏保护蛋白和细胞骨架蛋白增加,而心肌细胞收缩蛋白减少。这些对局部缺血的内在适应以牺牲收缩功能为代价维持了心肌细胞的长期活力。