Klein Eili Y, Serohijos Adrian W R, Choi Jeong-Mo, Shakhnovich Eugene I, Pekosz Andrew
Center for Advanced Modeling in the Social, Behavioral, and Health Sciences, Department of Emergency Medicine, Johns Hopkins School of Medicine, Baltimore, Maryland, United States of America; Center for Disease Dynamics, Economics, and Policy, Washington, DC, United States of America.
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts, United States of America.
PLoS One. 2014 Apr 3;9(4):e93632. doi: 10.1371/journal.pone.0093632. eCollection 2014.
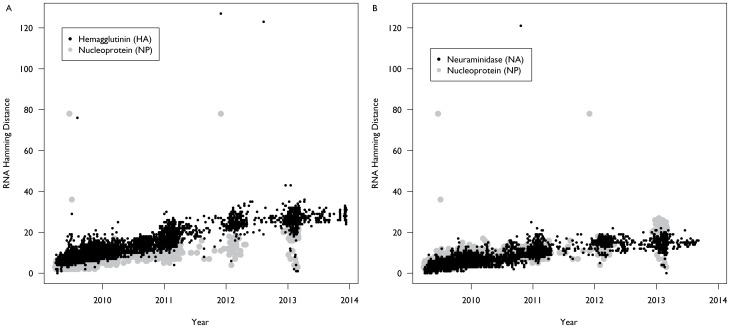
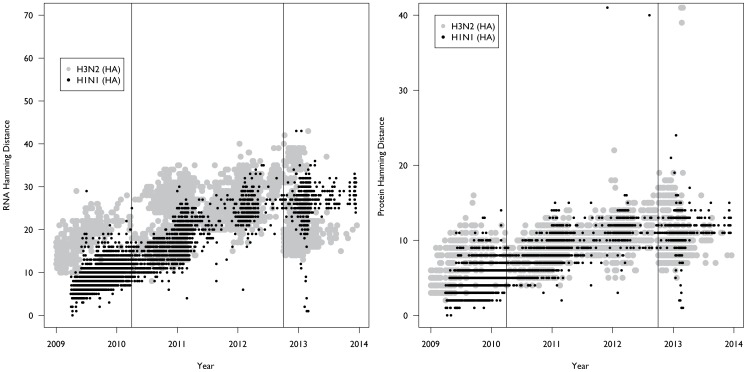
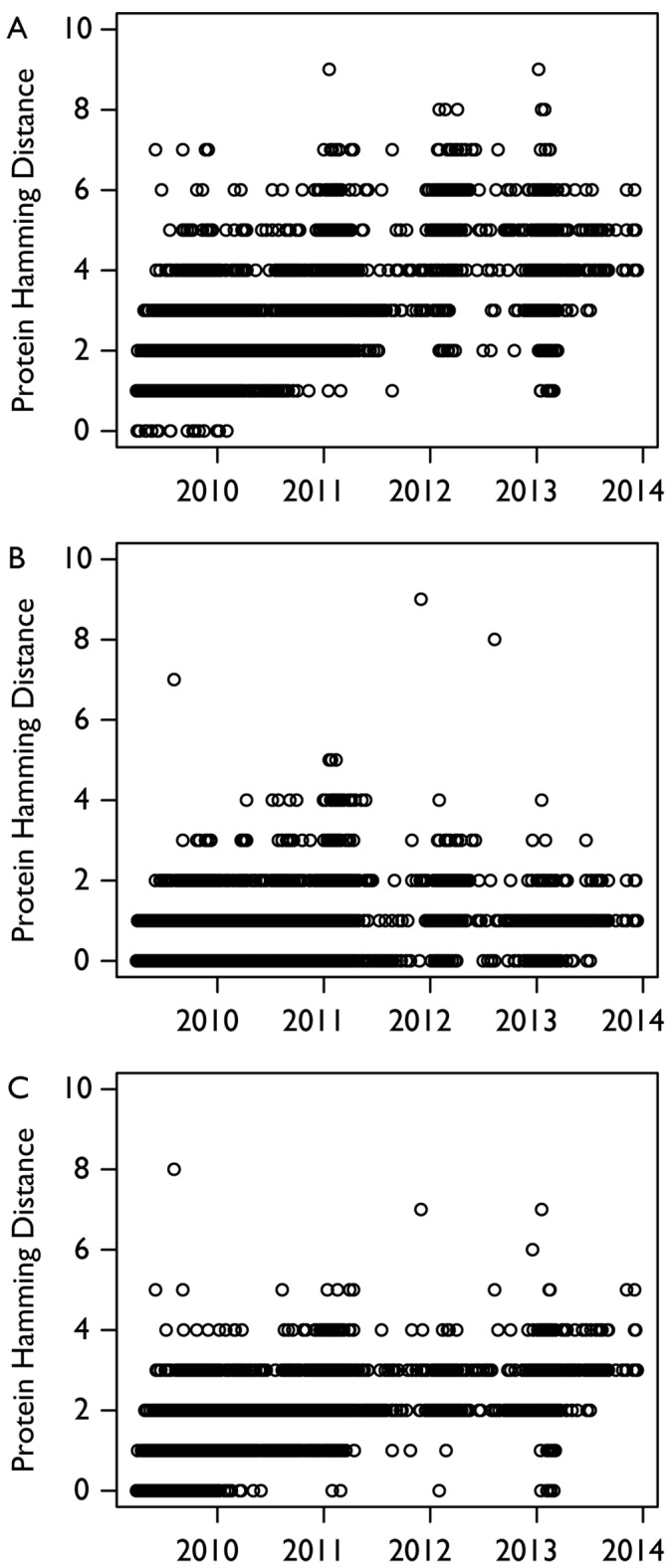
The emergence of a novel A(H1N1) strain in 2009 was the first influenza pandemic of the genomic age, and unprecedented surveillance of the virus provides the opportunity to better understand the evolution of influenza. We examined changes in the nucleotide coding regions and the amino acid sequences of the hemagglutinin (HA), neuraminidase (NA), and nucleoprotein (NP) segments of the A(H1N1)pdm09 strain using publicly available data. We calculated the nucleotide and amino acid hamming distance from the vaccine strain A/California/07/2009 for each sequence. We also estimated Pepitope-a measure of antigenic diversity based on changes in the epitope regions-for each isolate. Finally, we compared our results to A(H3N2) strains collected over the same period. Our analysis found that the mean hamming distance for the HA protein of the A(H1N1)pdm09 strain increased from 3.6 (standard deviation [SD]: 1.3) in 2009 to 11.7 (SD: 1.0) in 2013, while the mean hamming distance in the coding region increased from 7.4 (SD: 2.2) in 2009 to 28.3 (SD: 2.1) in 2013. These trends are broadly similar to the rate of mutation in H3N2 over the same time period. However, in contrast to H3N2 strains, the rate of mutation accumulation has slowed in recent years. Our results are notable because, over the course of the study, mutation rates in H3N2 similar to that seen with A(H1N1)pdm09 led to the emergence of two antigenic drift variants. However, while there has been an H1N1 epidemic in North America this season, evidence to date indicates the vaccine is still effective, suggesting the epidemic is not due to the emergence of an antigenic drift variant. Our results suggest that more research is needed to understand how viral mutations are related to vaccine effectiveness so that future vaccine choices and development can be more predictive.
2009年新型甲型H1N1流感病毒株的出现是基因组时代的首次流感大流行,对该病毒前所未有的监测为更好地了解流感的演变提供了契机。我们利用公开数据研究了甲型H1N1pdm09病毒株的血凝素(HA)、神经氨酸酶(NA)和核蛋白(NP)片段的核苷酸编码区及氨基酸序列的变化。我们计算了每个序列与疫苗株A/California/07/2009之间的核苷酸和氨基酸汉明距离。我们还估算了每个分离株的肽表位——基于表位区域变化的抗原多样性指标。最后,我们将结果与同期收集的甲型H3N2病毒株进行了比较。我们的分析发现,甲型H1N1pdm09病毒株HA蛋白的平均汉明距离从2009年的3.6(标准差[SD]:1.3)增加到2013年的11.7(SD:1.0),而编码区的平均汉明距离从2009年的7.4(SD:2.2)增加到2013年的28.3(SD:2.1)。这些趋势与同期H3N2的突变率大致相似。然而,与H3N2病毒株不同的是,近年来突变积累速率有所放缓。我们的结果值得关注,因为在研究过程中,与甲型H1N1pdm09类似的H3N2突变率导致了两种抗原漂移变异株的出现。然而,尽管北美本季度出现了H1N1疫情,但迄今为止的证据表明疫苗仍然有效,这表明此次疫情并非由抗原漂移变异株的出现所致。我们的结果表明,需要开展更多研究来了解病毒突变与疫苗效力之间的关系,以便未来的疫苗选择和研发更具预测性。