Renfrew P Douglas, Craven Timothy W, Butterfoss Glenn L, Kirshenbaum Kent, Bonneau Richard
Center for Genomics and Systems Biology, Department of Biology, ‡Department of Chemistry, and §Courant Institute of Mathematical Sciences, Computer Science Department, New York University , New York, New York 10003, United States.
J Am Chem Soc. 2014 Jun 18;136(24):8772-82. doi: 10.1021/ja503776z. Epub 2014 Jun 9.
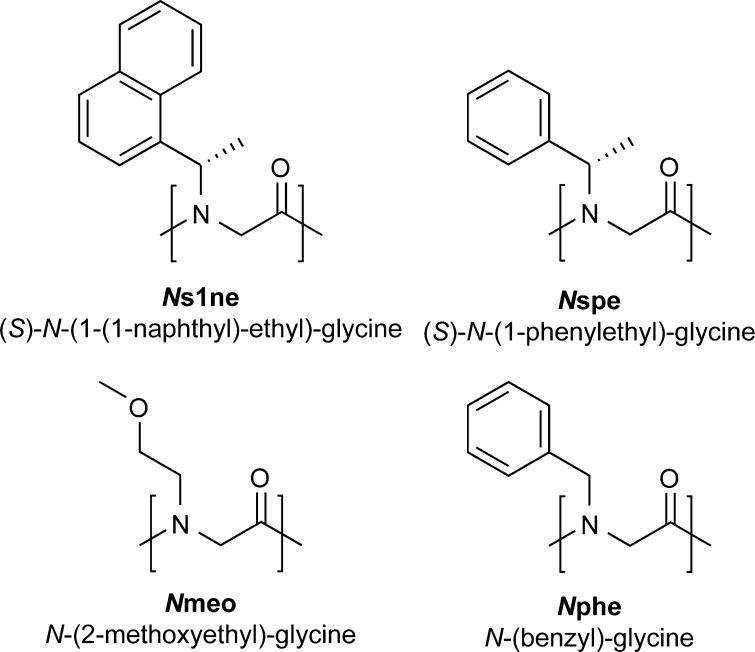
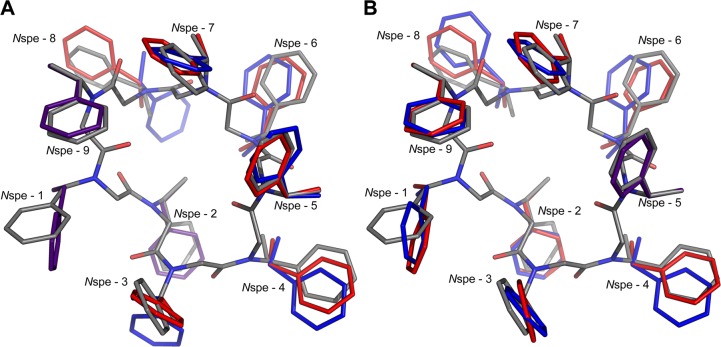
Peptoids are a family of synthetic oligomers composed of N-substituted glycine units. Along with other "foldamer" systems, peptoid oligomer sequences can be predictably designed to form a variety of stable secondary structures. It is not yet evident if foldamer design can be extended to reliably create tertiary structure features that mimic more complex biomolecular folds and functions. Computational modeling and prediction of peptoid conformations will likely play a critical role in enabling complex biomimetic designs. We introduce a computational approach to provide accurate conformational and energetic parameters for peptoid side chains needed for successful modeling and design. We find that peptoids can be described by a "rotamer" treatment, similar to that established for proteins, in which the peptoid side chains display rotational isomerism to populate discrete regions of the conformational landscape. Because of the insufficient number of solved peptoid structures, we have calculated the relative energies of side-chain conformational states to provide a backbone-dependent (BBD) rotamer library for a set of 54 different peptoid side chains. We evaluated two rotamer library development methods that employ quantum mechanics (QM) and/or molecular mechanics (MM) energy calculations to identify side-chain rotamers. We show by comparison to experimental peptoid structures that both methods provide an accurate prediction of peptoid side chain placements in folded peptoid oligomers and at protein interfaces. We have incorporated our peptoid rotamer libraries into ROSETTA, a molecular design package previously validated in the context of protein design and structure prediction.
类肽是由N-取代甘氨酸单元组成的一类合成低聚物。与其他“折叠体”系统一样,类肽低聚物序列可以通过可预测的设计形成多种稳定的二级结构。折叠体设计是否能够扩展到可靠地创建模仿更复杂生物分子折叠和功能的三级结构特征,目前尚不清楚。类肽构象的计算建模和预测可能在实现复杂的仿生设计中发挥关键作用。我们引入了一种计算方法,为成功建模和设计所需的类肽侧链提供准确的构象和能量参数。我们发现类肽可以通过一种“旋转异构体”处理来描述,类似于蛋白质中确立的方法,其中类肽侧链表现出旋转异构现象,以占据构象空间的离散区域。由于已解析的类肽结构数量不足,我们计算了侧链构象状态的相对能量,为一组54种不同的类肽侧链提供了一个依赖于主链的(BBD)旋转异构体文库。我们评估了两种利用量子力学(QM)和/或分子力学(MM)能量计算来识别侧链旋转异构体的旋转异构体文库开发方法。通过与实验类肽结构的比较,我们表明这两种方法都能准确预测折叠的类肽低聚物和蛋白质界面中类肽侧链的位置。我们已将我们的类肽旋转异构体文库整合到ROSETTA中,ROSETTA是一个先前在蛋白质设计和结构预测背景下经过验证的分子设计软件包。