Kalogeris Theodore J, Baines Christopher, Korthuis Ronald J
Department of Medical Pharmacology and Physiology, University of Missouri, Columbia, Missouri, United States of America.
Department of Biomedical Sciences, University of Missouri, Columbia, Missouri, United States of America; Dalton Cardiovascular Research Center, University of Missouri, Columbia, Missouri, United States of America.
PLoS One. 2014 Jun 10;9(6):e98459. doi: 10.1371/journal.pone.0098459. eCollection 2014.
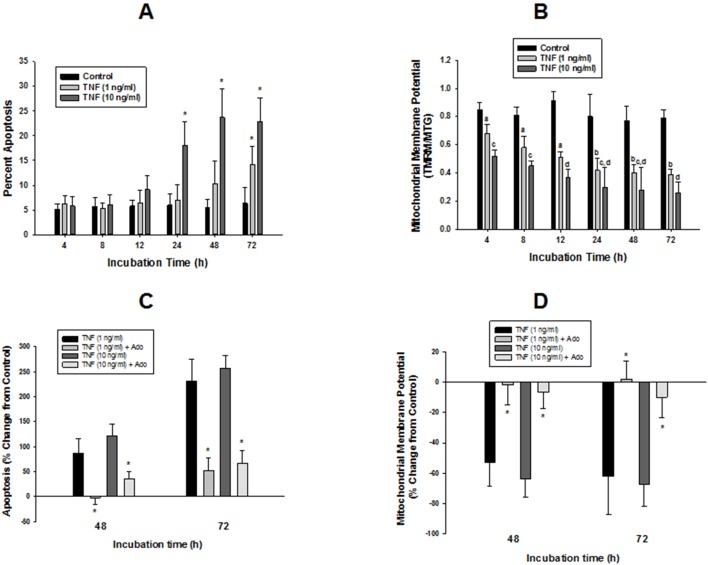
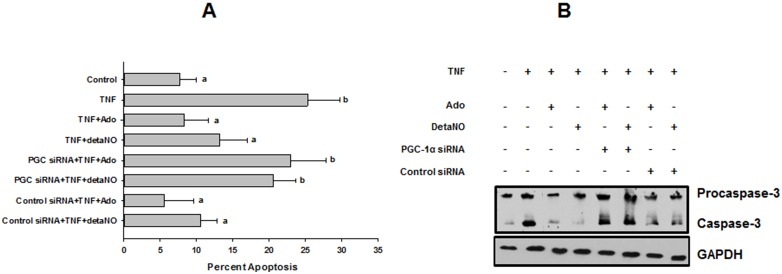
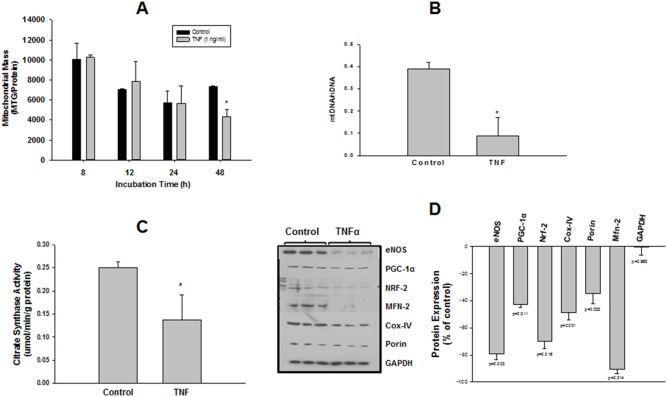
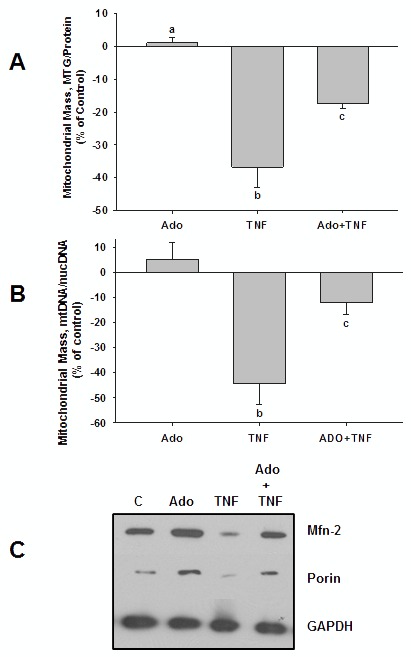
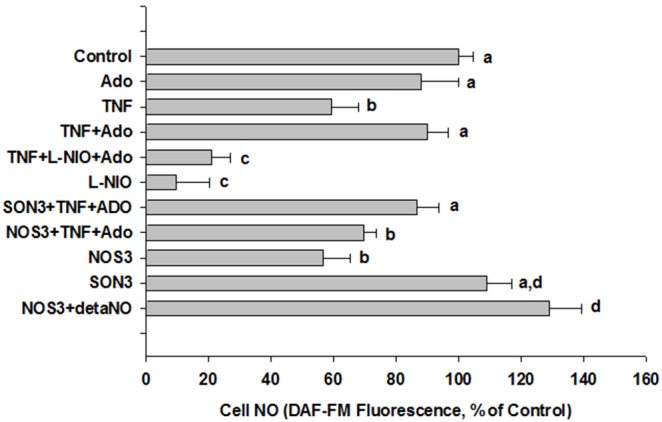
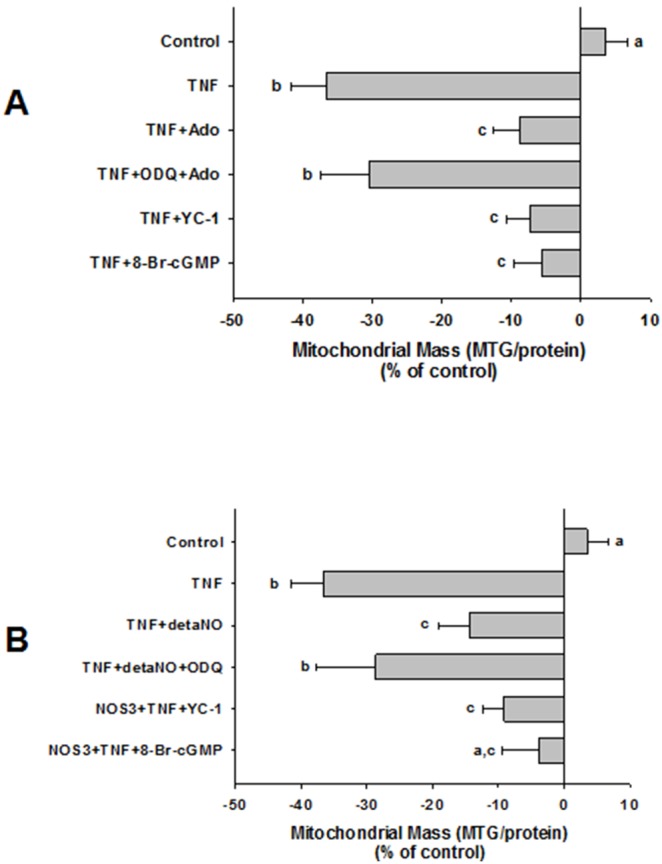
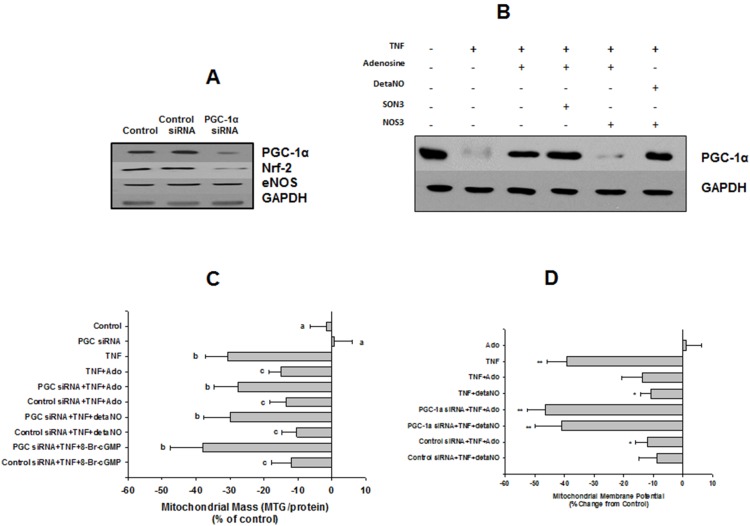
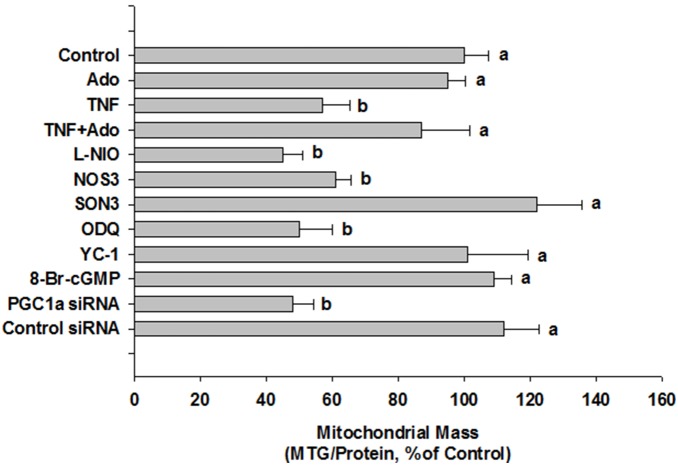
We tested whether adenosine, a cytoprotective mediator and trigger of preconditioning, could protect endothelial cells from inflammation-induced deficits in mitochondrial biogenesis and function. We examined this question using human microvascular endothelial cells exposed to TNFα. TNFα produced time and dose-dependent decreases in mitochondrial membrane potential, cellular ATP levels, and mitochondrial mass, preceding an increase in apoptosis. These effects were prevented by co-incubation with adenosine, a nitric oxide (NO) donor, a guanylate cyclase (GC) activator, or a cell-permeant cyclic GMP (cGMP) analog. The effects of adenosine were blocked by a nitric oxide synthase inhibitor, a soluble guanylate cyclase inhibitor, a morpholino antisense oligonucleotide to endothelial nitric oxide synthase (eNOS), or siRNA knockdown of the transcriptional coactivator, PGC-1α. Incubation with exogenous NO, a GC activator, or a cGMP analog reversed the effect of eNOS knockdown, while the effect of NO was blocked by inhibition of GC. The protective effects of NO and cGMP analog were prevented by siRNA to PGC-1α. TNFα also decreased expression of eNOS, cellular NO levels, and PGC-1α expression, which were reversed by adenosine. Exogenous NO, but not adenosine, rescued expression of PGC-1α in cells in which eNOS expression was knocked down by eNOS antisense treatment. Thus, TNFα elicits decreases in endothelial mitochondrial function and mass, and an increase in apoptosis. These effects were reversed by adenosine, an effect mediated by eNOS-synthesized NO, acting via soluble guanylate cyclase/cGMP to activate a mitochondrial biogenesis regulatory program under the control of PGC-1α. These results support the existence of an adenosine-triggered, mito-and cytoprotective mechanism dependent upon an eNOS-PGC-1α regulatory pathway, which acts to preserve endothelial mitochondrial function and mass during inflammatory challenge.
我们测试了作为细胞保护介质和预处理触发因子的腺苷是否能够保护内皮细胞免受炎症诱导的线粒体生物合成及功能缺陷的影响。我们使用暴露于肿瘤坏死因子α(TNFα)的人微血管内皮细胞来研究这一问题。TNFα导致线粒体膜电位、细胞ATP水平和线粒体质量呈时间和剂量依赖性下降,随后细胞凋亡增加。与腺苷、一氧化氮(NO)供体、鸟苷酸环化酶(GC)激活剂或细胞渗透性环鸟苷酸(cGMP)类似物共同孵育可防止这些效应。腺苷的作用被一氧化氮合酶抑制剂、可溶性鸟苷酸环化酶抑制剂、针对内皮型一氧化氮合酶(eNOS)的吗啉代反义寡核苷酸或转录共激活因子PGC-1α的小干扰RNA(siRNA)敲低所阻断。用外源性NO、GC激活剂或cGMP类似物孵育可逆转eNOS敲低的效应,而NO的效应被GC抑制所阻断。NO和cGMP类似物的保护作用被PGC-1α的siRNA所阻止。TNFα还降低了eNOS的表达、细胞内NO水平和PGC-1α的表达,而腺苷可使其逆转。外源性NO而非腺苷可挽救经eNOS反义处理敲低eNOS表达的细胞中PGC-1α的表达。因此,TNFα导致内皮线粒体功能和质量下降以及细胞凋亡增加。这些效应被腺苷逆转,该效应由eNOS合成的NO介导,通过可溶性鸟苷酸环化酶/cGMP起作用,以激活在PGC-1α控制下的线粒体生物合成调节程序。这些结果支持存在一种依赖于eNOS-PGC-1α调节途径的腺苷触发的线粒体和细胞保护机制,该机制在炎症刺激期间起到保护内皮线粒体功能和质量的作用。