Wertheim Joel O, Smith Martin D, Smith Davey M, Scheffler Konrad, Kosakovsky Pond Sergei L
Department of Medicine, University of California, San Diego
Bioinformatics and Systems Biology Graduate Program, University of California, San Diego.
Mol Biol Evol. 2014 Sep;31(9):2356-64. doi: 10.1093/molbev/msu185. Epub 2014 Jun 10.
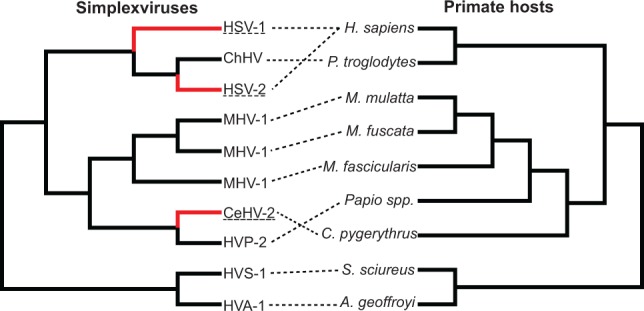
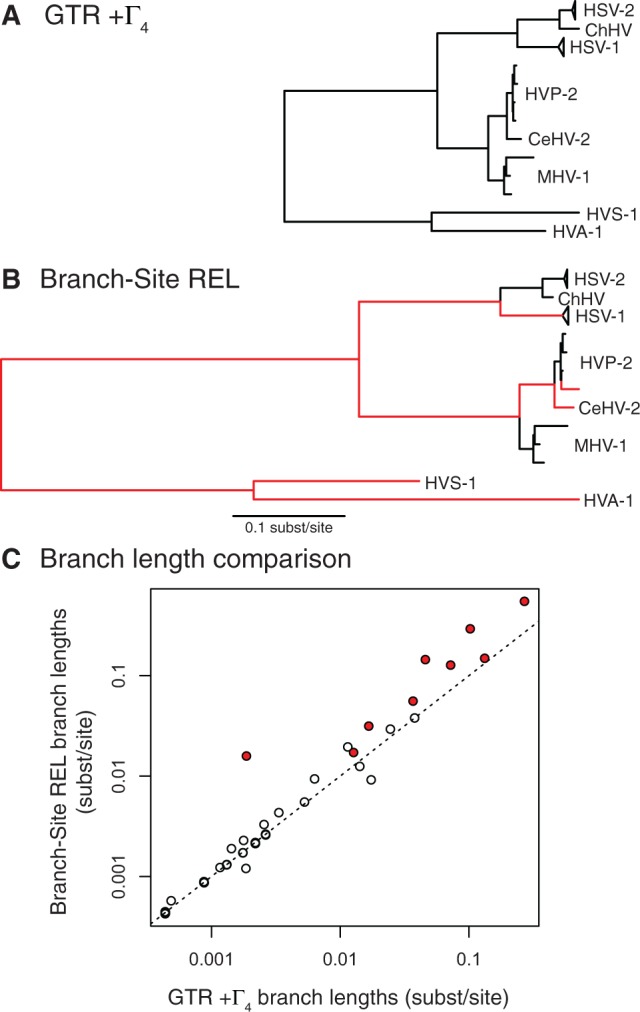
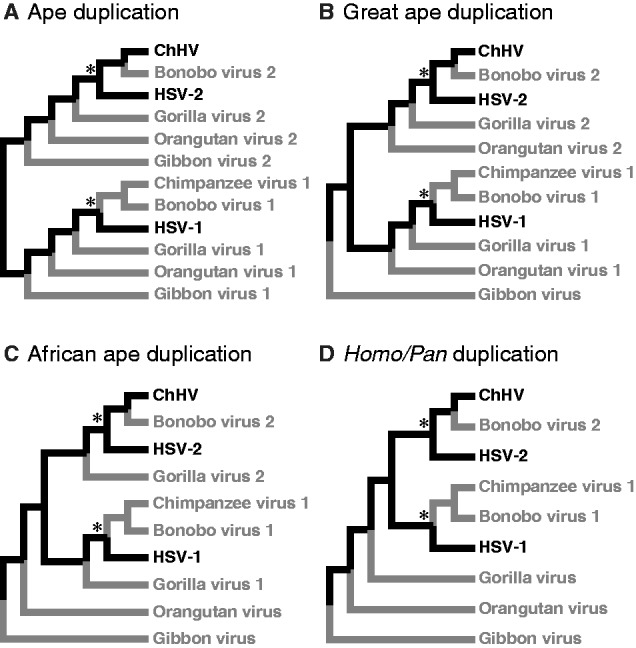
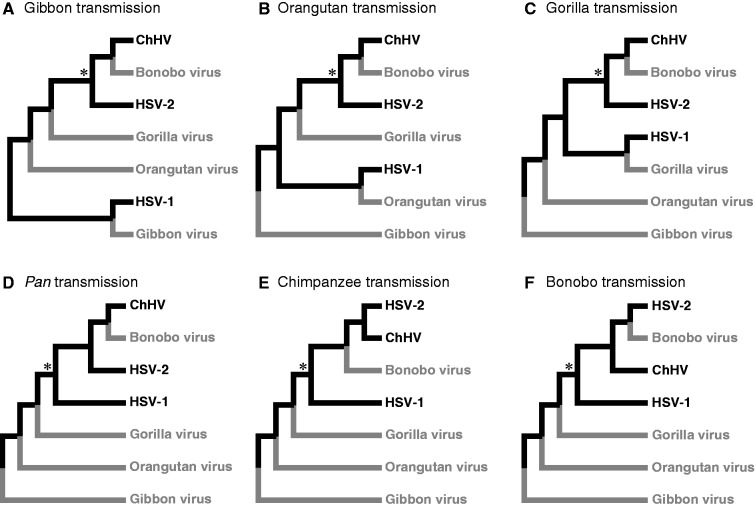
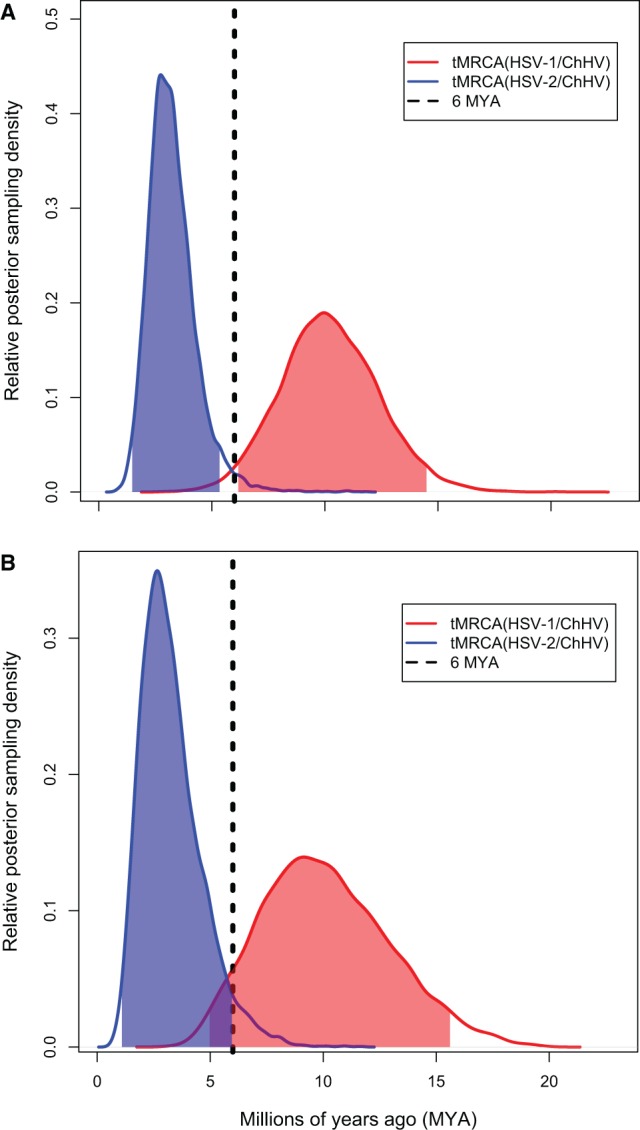
Herpesviruses have been infecting and codiverging with their vertebrate hosts for hundreds of millions of years. The primate simplex viruses exemplify this pattern of virus-host codivergence, at a minimum, as far back as the most recent common ancestor of New World monkeys, Old World monkeys, and apes. Humans are the only primate species known to be infected with two distinct herpes simplex viruses: HSV-1 and HSV-2. Human herpes simplex viruses are ubiquitous, with over two-thirds of the human population infected by at least one virus. Here, we investigated whether the additional human simplex virus is the result of ancient viral lineage duplication or cross-species transmission. We found that standard phylogenetic models of nucleotide substitution are inadequate for distinguishing among these competing hypotheses; the extent of synonymous substitutions causes a substantial underestimation of the lengths of some of the branches in the phylogeny, consistent with observations in other viruses (e.g., avian influenza, Ebola, and coronaviruses). To more accurately estimate ancient viral divergence times, we applied a branch-site random effects likelihood model of molecular evolution that allows the strength of natural selection to vary across both the viral phylogeny and the gene alignment. This selection-informed model favored a scenario in which HSV-1 is the result of ancient codivergence and HSV-2 arose from a cross-species transmission event from the ancestor of modern chimpanzees to an extinct Homo precursor of modern humans, around 1.6 Ma. These results provide a new framework for understanding human herpes simplex virus evolution and demonstrate the importance of using selection-informed models of sequence evolution when investigating viral origin hypotheses.
疱疹病毒已经感染脊椎动物宿主并与之共同进化了数亿年。灵长类单纯疱疹病毒至少从新世界猴、旧世界猴和猿的最近共同祖先开始就例证了这种病毒 - 宿主共同进化模式。人类是已知感染两种不同单纯疱疹病毒(HSV - 1和HSV - 2)的唯一灵长类物种。人类单纯疱疹病毒无处不在,超过三分之二的人类至少感染了一种病毒。在这里,我们研究了额外的人类单纯疱疹病毒是古代病毒谱系复制还是跨物种传播的结果。我们发现,标准的核苷酸替换系统发育模型不足以区分这些相互竞争的假说;同义替换的程度导致系统发育中某些分支长度的严重低估,这与其他病毒(如禽流感、埃博拉和冠状病毒)的观察结果一致。为了更准确地估计古代病毒的分歧时间,我们应用了一种分子进化的分支位点随机效应似然模型,该模型允许自然选择的强度在病毒系统发育和基因比对中变化。这种考虑选择因素的模型支持一种假说,即HSV - 1是古代共同进化的结果,而HSV - 2则起源于大约160万年前从现代黑猩猩的祖先到现代人类已灭绝的智人前身的跨物种传播事件。这些结果为理解人类单纯疱疹病毒的进化提供了一个新框架,并证明了在研究病毒起源假说时使用考虑选择因素的序列进化模型的重要性。