Chen Yi-An, Tripathi Lokesh P, Dessailly Benoit H, Nyström-Persson Johan, Ahmad Shandar, Mizuguchi Kenji
National Institute of Biomedical Innovation, Ibaraki, Osaka, Japan.
PLoS One. 2014 Jun 11;9(6):e99030. doi: 10.1371/journal.pone.0099030. eCollection 2014.
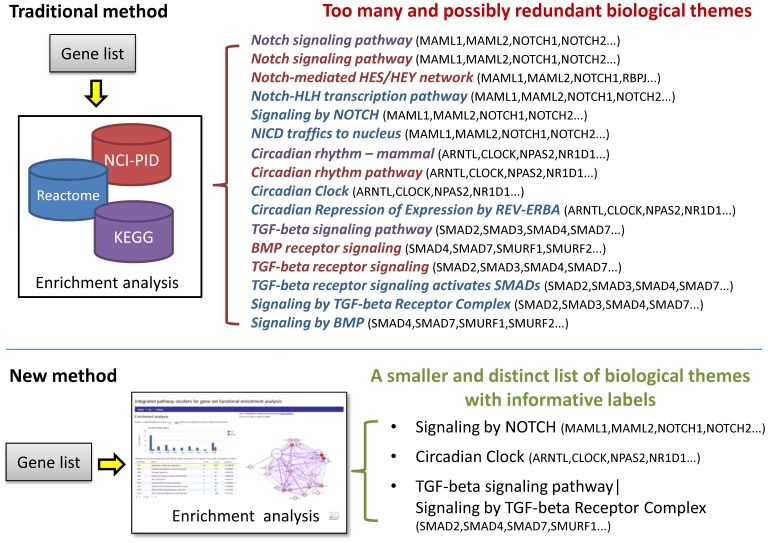
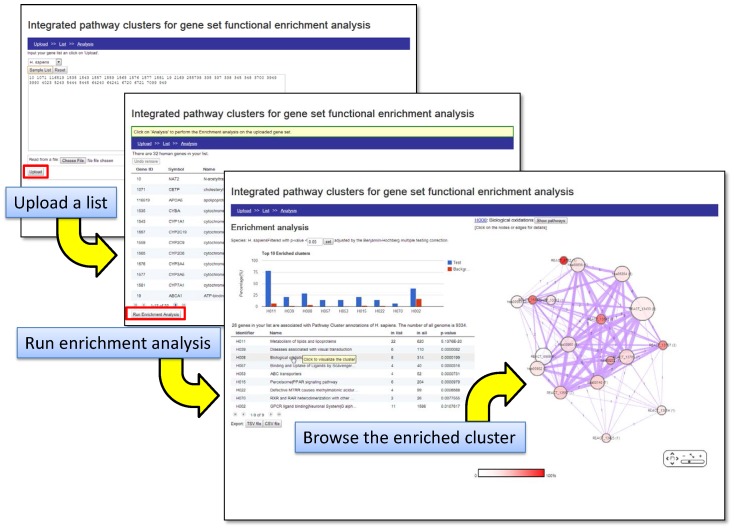
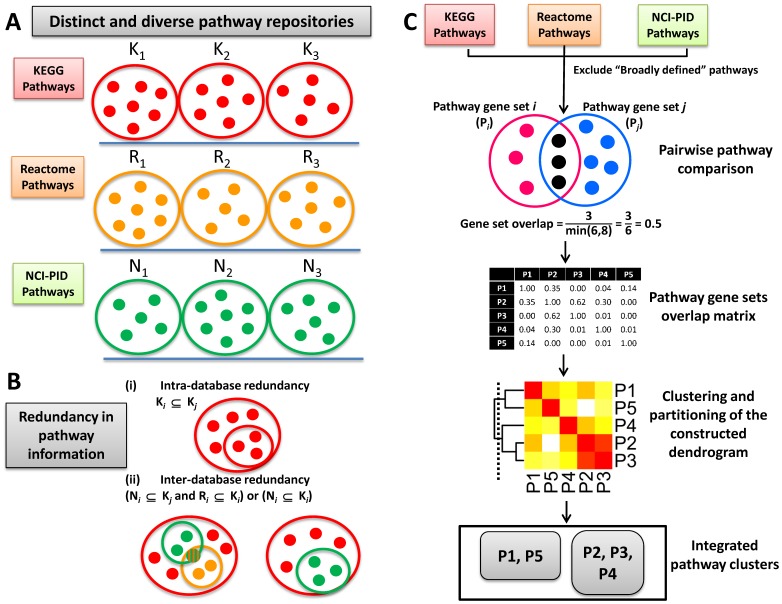
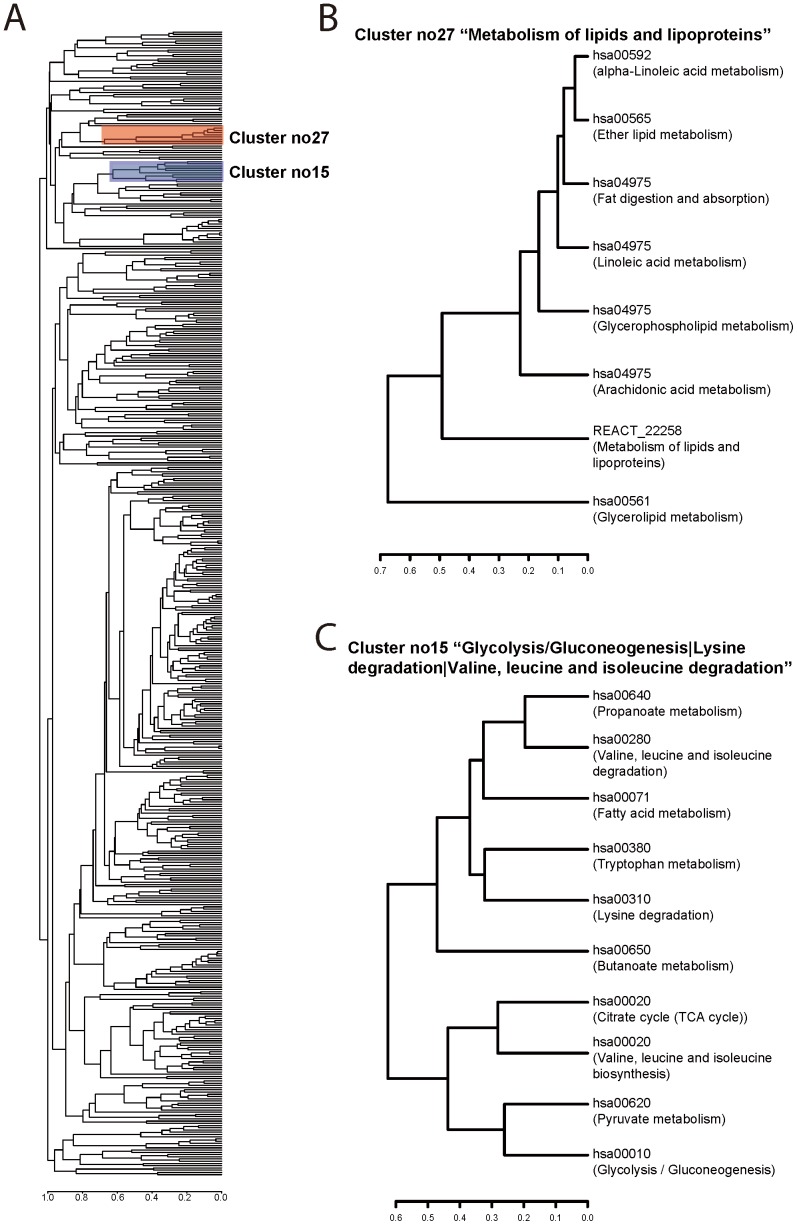
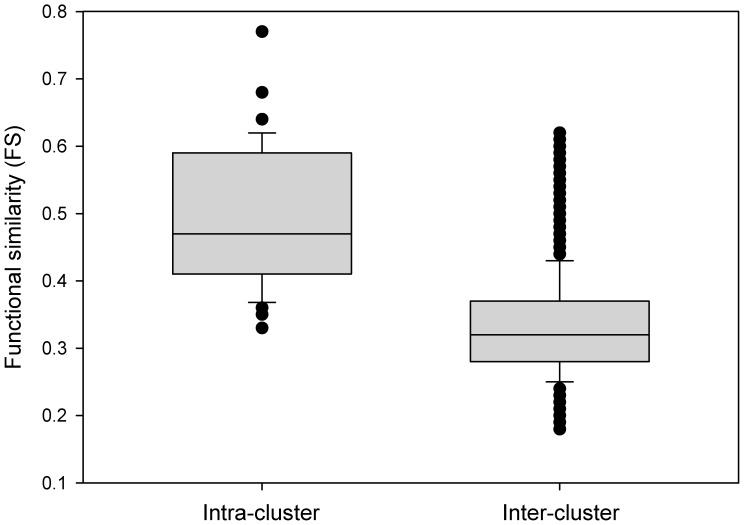
Prioritising candidate genes for further experimental characterisation is an essential, yet challenging task in biomedical research. One way of achieving this goal is to identify specific biological themes that are enriched within the gene set of interest to obtain insights into the biological phenomena under study. Biological pathway data have been particularly useful in identifying functional associations of genes and/or gene sets. However, biological pathway information as compiled in varied repositories often differs in scope and content, preventing a more effective and comprehensive characterisation of gene sets. Here we describe a new approach to constructing biologically coherent gene sets from pathway data in major public repositories and employing them for functional analysis of large gene sets. We first revealed significant overlaps in gene content between different pathways and then defined a clustering method based on the shared gene content and the similarity of gene overlap patterns. We established the biological relevance of the constructed pathway clusters using independent quantitative measures and we finally demonstrated the effectiveness of the constructed pathway clusters in comparative functional enrichment analysis of gene sets associated with diverse human diseases gathered from the literature. The pathway clusters and gene mappings have been integrated into the TargetMine data warehouse and are likely to provide a concise, manageable and biologically relevant means of functional analysis of gene sets and to facilitate candidate gene prioritisation.
在生物医学研究中,确定用于进一步实验表征的候选基因是一项至关重要但又具有挑战性的任务。实现这一目标的一种方法是识别在感兴趣的基因集中富集的特定生物学主题,以深入了解所研究的生物学现象。生物通路数据在识别基因和/或基因集的功能关联方面特别有用。然而,不同数据库中汇编的生物通路信息在范围和内容上往往存在差异,这阻碍了对基因集进行更有效和全面的表征。在此,我们描述了一种新方法,该方法从主要公共数据库中的通路数据构建生物学上连贯的基因集,并将其用于大型基因集的功能分析。我们首先揭示了不同通路之间基因内容的显著重叠,然后基于共享的基因内容和基因重叠模式的相似性定义了一种聚类方法。我们使用独立的定量指标确定了所构建通路簇的生物学相关性,最后证明了所构建通路簇在对从文献中收集的与多种人类疾病相关的基因集进行比较功能富集分析中的有效性。通路簇和基因映射已整合到TargetMine数据仓库中,可能会为基因集的功能分析提供一种简洁、可管理且具有生物学相关性的方法,并有助于候选基因的优先级排序。