Oliveira Saulo H P, Ferraz Felipe A N, Honorato Rodrigo V, Xavier-Neto José, Sobreira Tiago J P, de Oliveira Paulo S L
National Laboratory of Biosciences, P,O, Box 6192, CEP 13083-970 Campinas, SP, Brazil.
BMC Bioinformatics. 2014 Jun 17;15:197. doi: 10.1186/1471-2105-15-197.
The characterization of protein binding sites is a major challenge in computational biology. Proteins interact with a wide variety of molecules and understanding of such complex interactions is essential to gain deeper knowledge of protein function. Shape complementarity is known to be important in determining protein-ligand interactions. Furthermore, these protein structural features have been shown to be useful in assisting medicinal chemists during lead discovery and optimization.
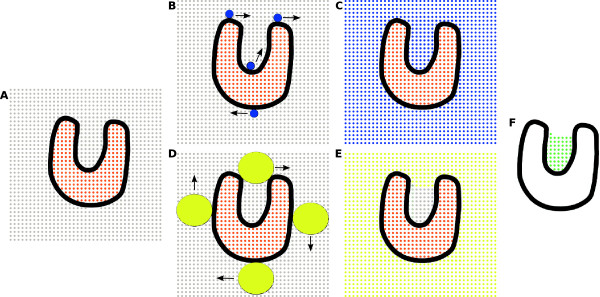
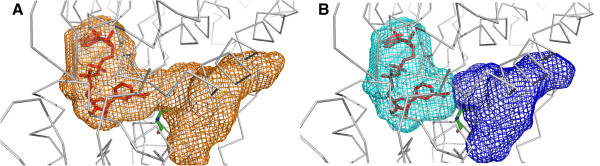
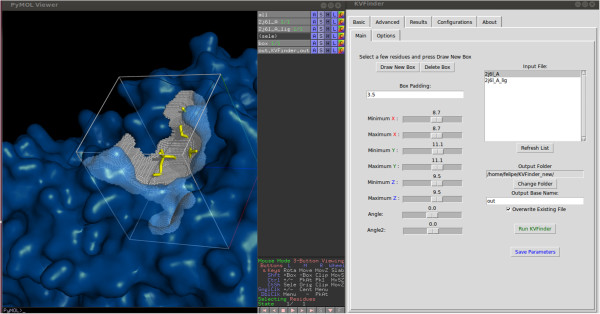
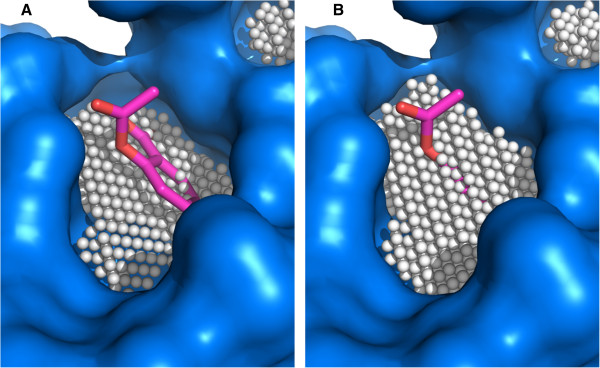
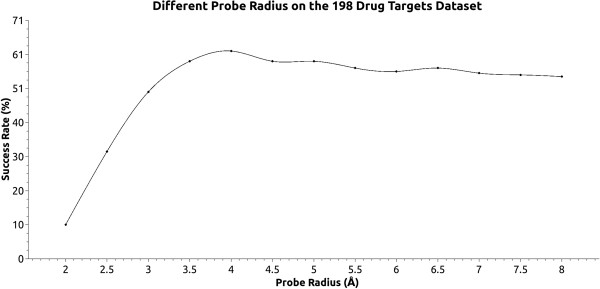
We developed KVFinder, a highly versatile and easy-to-use tool for cavity prospection and spatial characterization. KVFinder is a geometry-based method that has an innovative customization of the search space. This feature provides the possibility of cavity segmentation, which alongside with the large set of customizable parameters, allows detailed cavity analyses. Although the main focus of KVFinder is the steered prospection of cavities, we tested it against a benchmark dataset of 198 known drug targets in order to validate our software and compare it with some of the largely accepted methods. Using the one click mode, we performed better than most of the other methods, staying behind only of hybrid prospection methods. When using just one of KVFinder's customizable features, we were able to outperform all other compared methods. KVFinder is also user friendly, as it is available as a PyMOL plugin, or command-line version.
KVFinder presents novel usability features, granting full customizable and highly detailed cavity prospection on proteins, alongside with a friendly graphical interface. KVFinder is freely available on http://lnbio.cnpem.br/bioinformatics/main/software/.
蛋白质结合位点的表征是计算生物学中的一项重大挑战。蛋白质与各种各样的分子相互作用,理解这种复杂的相互作用对于深入了解蛋白质功能至关重要。已知形状互补性在确定蛋白质 - 配体相互作用中很重要。此外,这些蛋白质结构特征已被证明在先导化合物发现和优化过程中对药物化学家很有用。
我们开发了KVFinder,这是一种用于腔探测和空间表征的高度通用且易于使用的工具。KVFinder是一种基于几何的方法,对搜索空间进行了创新定制。此功能提供了腔分割的可能性,与大量可定制参数一起,允许进行详细的腔分析。尽管KVFinder的主要重点是腔的定向探测,但我们针对198个已知药物靶点的基准数据集对其进行了测试,以验证我们的软件并将其与一些广泛接受的方法进行比较。使用一键模式,我们的表现优于大多数其他方法,仅落后于混合探测方法。当仅使用KVFinder的一个可定制功能时,我们能够超越所有其他比较方法。KVFinder也很用户友好,因为它可以作为PyMOL插件或命令行版本使用。
KVFinder具有新颖的可用性特征,可对蛋白质进行完全可定制且高度详细的腔探测,同时具有友好的图形界面。KVFinder可在http://lnbio.cnpem.br/bioinformatics/main/software/上免费获取。